

AMPK alterations in cardiac physiology and pathology: enemy or ally?
- PMID: 16690706
- PMCID: PMC1817803
- DOI: 10.1113/jphysiol.2006.109389
AMPK alterations in cardiac physiology and pathology: enemy or ally?
Abstract
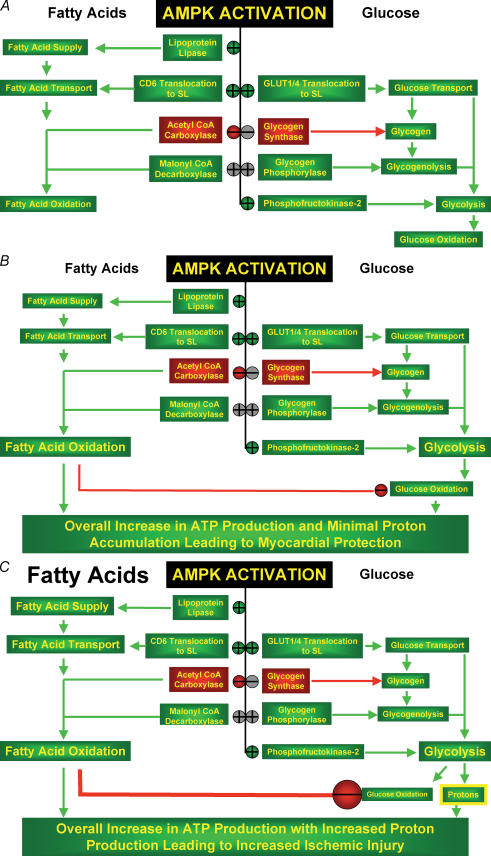
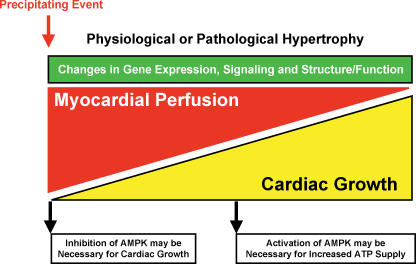
AMP-activated protein kinase (AMPK) has emerged as a key regulator of energy metabolism in the heart. The high energy demands of the heart are primarily met by the metabolism of both fatty acids and glucose, both processes being regulated by AMPK. During myocardial ischaemia a rapid activation of AMPK occurs, resulting in an activation of both glucose uptake and glycolysis, as well as an increase in fatty acid oxidation. This activation of AMPK has the potential to increase energy production and to inhibit apoptosis, thereby protecting the heart during the ischaemic stress. However, at clinically relevant high levels of fatty acids, ischaemic-induced activation of AMPK also stimulates fatty acid oxidation during and following ischaemia. This can contribute to ischaemic injury secondary to an inhibition of glucose oxidation, which results in a decrease in cardiac efficiency. In a number of other non-cardiac tissues, AMPK has been shown to have pro-apoptotic effects. As a result, the question of whether AMPK activation benefits or harms the ischaemic heart remains controversial. The role of AMPK in cardiac hypertrophy is also controversial. Activation of AMPK inhibits protein synthesis, and may be an adaptive response to pathological cardiac hypertrophy. However, none of mouse models of AMPK deficiency (excluding those that may involve the gamma2 subunit mutations) demonstrate increased cardiac mass, suggesting that AMPK is not essential for restriction of cardiac growth. In addition to the potential effects of AMPK on myofibrillar hypertrophy associated with pressure overload, there is also controversy with respect to the cardiac hypertrophy associated with the gamma2 subunit mutations. In the cardiac hypertrophy associated with glycogen overload, both activating and inactivating mutations of AMPK in mice are associated with a marked cardiac hypertrophy. This review will address the issue of whether AMPK activation acts as an enemy or ally to the ischaemic and hypertrophied heart. Resolving this issue has important implications as to whether therapeutic approaches to protect the ischaemic heart should be developed which either activate or inhibit AMPK.
Figures

References
-
- Ahmad F, Arad M, Musi N, He H, Wolf C, Branco D, Perez-Atayde AR, Stapleton D, Bali D, Xing Y, Tian R, Goodyear LJ, Berul CI, Ingwall JS, Seidman CE, Seidman JG. Increased α2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. 2005;112:3140–3148. - PubMed
-
- Allard MF, Flint JD, English JC, Henning SL, Salamanca MC, Kamimura CT, English DR. Calcium overload during reperfusion is accelerated in isolated hypertrophied rat hearts. J Mol Cell Cardiol. 1994a;26:1551–1563. - PubMed
-
- Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994b;267:H742–H750. - PubMed
-
- Allard MF, Wambolt RB, Longnus SL, Grist M, Lydell CP, Parsons HL, Rodrigues B, Hall JL, Stanley WC, Bondy GP. Hypertrophied rat hearts are less responsive to the metabolic and functional effects of insulin. Am J Physiol Endocrinol Metab. 2000;279:E487–E493. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous