

Prenatal diagnosis of myopathy, encephalopathy, lactic acidosis, and stroke-like syndrome: contribution to understanding mitochondrial DNA segregation during human embryofetal development
- PMID: 16690729
- PMCID: PMC2563165
- DOI: 10.1136/jmg.2005.034140
Prenatal diagnosis of myopathy, encephalopathy, lactic acidosis, and stroke-like syndrome: contribution to understanding mitochondrial DNA segregation during human embryofetal development
Abstract
Introduction: Myopathy, encephalopathy, lactic acidosis, and stroke-like (MELAS) syndrome, a maternally inherited disorder that is among the most common mitochondrial DNA (mtDNA) diseases, is usually associated with the m.3242A>G mutation of the mitochondrial tRNA(leu) gene. Very few data are available with respect to prenatal diagnosis of this serious disease. The rate of mutant versus wild-type mtDNA (heteroplasmy) in fetal DNA is indeed considered to be a poor indicator of postnatal outcome.
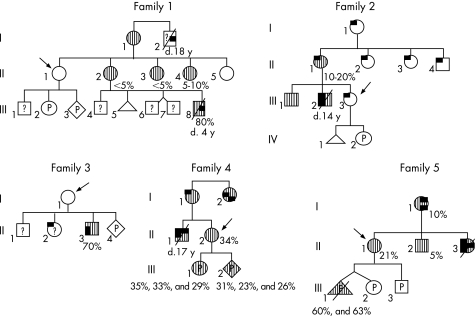
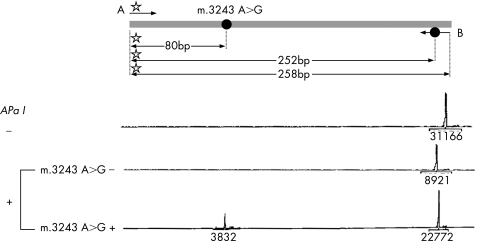
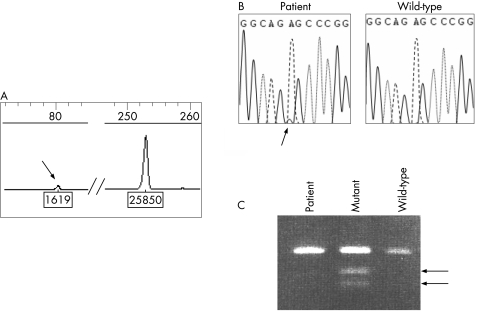
Materials and methods: Taking advantage of a novel semi-quantitative polymerase chain reaction test for m.3243A>G mutant load assessment, we carried out nine prenatal diagnoses in five unrelated women, using two different fetal tissues (chorionic villi v amniocytes) sampled at two or three different stages of pregnancy.
Results: Two of the five women, although not carrying m.3243A>G in blood or extra-blood tissues, were, however, considered at risk for transmission of the mutation, as they were closely related to MELAS-affected individuals. The absence of 3243A>G in the blood of first degree relatives was associated with no mutated mtDNA in the cardiovascular system (CVS) or amniocytes, and their three children are healthy, with a follow-up of 3 months-3 years. Among the six fetuses from the three carrier women, three were shown to be homoplasmic (0% mutant load), the remaining three being heteroplasmic, with a mutant load ranging from 23% to 63%. The fetal mutant load was fairly stable at two or three different stages of pregnancy in CVS and amniocytes. Although pregnancy was terminated in the case of the fetus with a 63% mutant load, all other children are healthy with a follow-up of 3 months-6 years.
Conclusion: These data suggest that a prenatal diagnosis for MELAS syndrome might be helpful for at-risk families.
Conflict of interest statement
Competing interests: None declared.
References
-
- Goto Y, Nonaka I, Horai S. A mutation in the tRNA (Leu) (UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990348651–653. - PubMed
-
- Ciafaloni E, Ricci E, Shanske S, Moraes C, Silvestri G, Hirano M, Simonetti S, Angelini C, Donati M A, Garcia C. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol 199231391–398. - PubMed
-
- Manouvrier S, Rotig A, Hannebique G, Gheerbrandt J D, Royer‐Legrain G, Munnich A, Parent M, Grunfeld J P, Largilliere C, Lombes A, Bonnefont J P. Point mutation of the mitochondrial tRNA(Leu) gene (A 3243 G) in maternally inherited hypertrophic cardiomyopathy, diabetes mellitus, renal failure, and sensorineural deafness. J Med Genet 199532654–656. - PMC - PubMed
-
- Poulton J, Marchington D. Progress in genetic counseling and prenatal diagnosis of maternally inherited mtDNA diseases. Neuromuscul Disord 200010484–487. - PubMed
-
- Chou Y J, Ou C Y, Hsu T Y, Liou C W, Lee C F, Tso D J, Wei Y H. Prenatal diagnosis of a fetus harboring an intermediate load of the A3243G mtDNA mutation in a maternal carrier diagnosed with MELAS syndrome. Prenat Diagn 200424367–370. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical