

The remarkable transport mechanism of P-glycoprotein: a multidrug transporter
- PMID: 16691488
- PMCID: PMC1459968
- DOI: 10.1007/s10863-005-9497-5
The remarkable transport mechanism of P-glycoprotein: a multidrug transporter
Abstract
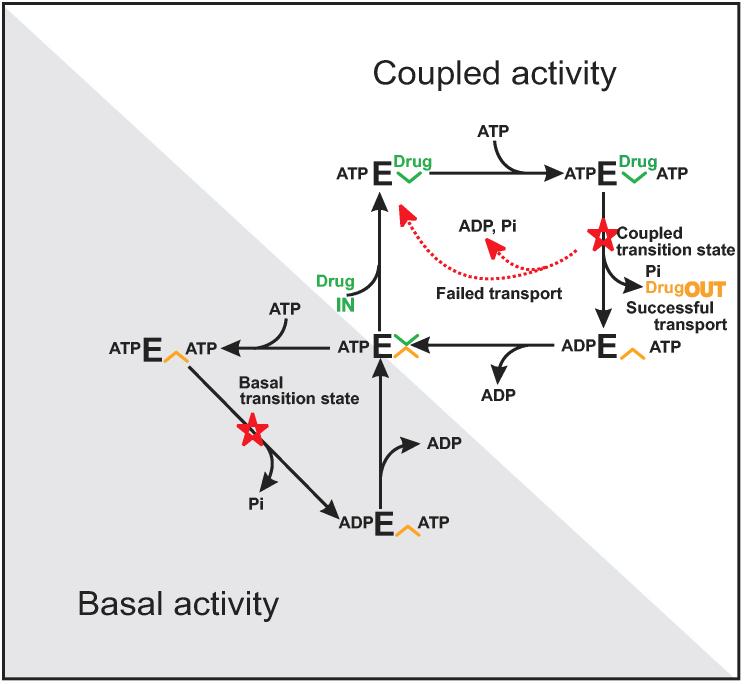
Human P-glycoprotein (ABCB1) is a primary multidrug transporter located in plasma membranes, that utilizes the energy of ATP hydrolysis to pump toxic xenobiotics out of cells. P-glycoprotein employs a most unusual molecular mechanism to perform this drug transport function. Here we review our work to elucidate the molecular mechanism of drug transport by P-glycoprotein. High level heterologous expression of human P-glycoprotein, in the yeast Saccharomyces cerevisiae, has facilitated biophysical studies in purified proteoliposome preparations. Development of novel spin-labeled transport substrates has allowed for quantitative and rigorous measurements of drug transport in real time by EPR spectroscopy. We have developed a new drug transport model of P-glycoprotein from the results of mutagenic, quantitative thermodynamic and kinetic studies. This model satisfactorily accounts for most of the unusual kinetic, coupling, and physiological features of P-glycoprotein. Additionally, an atomic detail structural model of P-glycoprotein has been devised to place our results within a proper structural context.
Figures

References
-
- Al-Shawi MK, Polar MK, Omote H, Figler RA. J. Biol. Chem. 2003;278:52629–52640. - PubMed
-
- Al-Shawi MK, Senior AE. J. Biol. Chem. 1993;268:4197–4206. - PubMed
-
- Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Annu. Rev. Pharmacol. Toxicol. 1999;39:361–398. - PubMed
-
- Chang G. J. Mol. Biol. 2003;330:419–430. - PubMed
-
- Chang G, Roth CB. Science. 2001;293:1793–1800. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
