

From the similarity analysis of protein cavities to the functional classification of protein families using cavbase
- PMID: 16697007
- PMCID: PMC7094329
- DOI: 10.1016/j.jmb.2006.04.024
From the similarity analysis of protein cavities to the functional classification of protein families using cavbase
Abstract
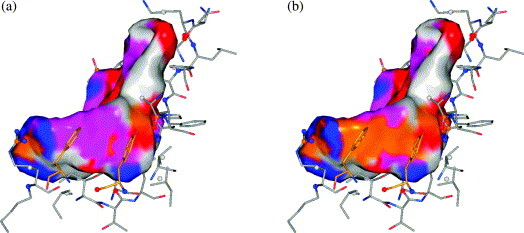
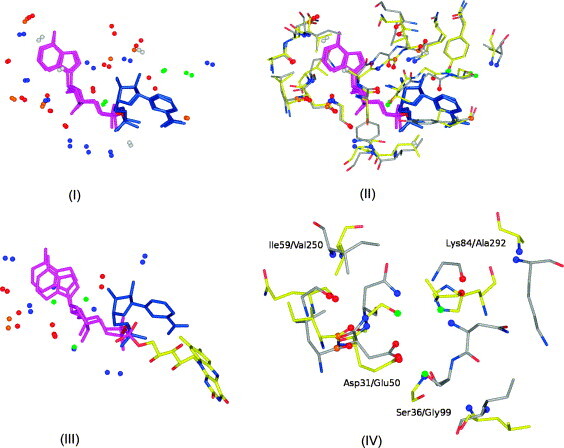
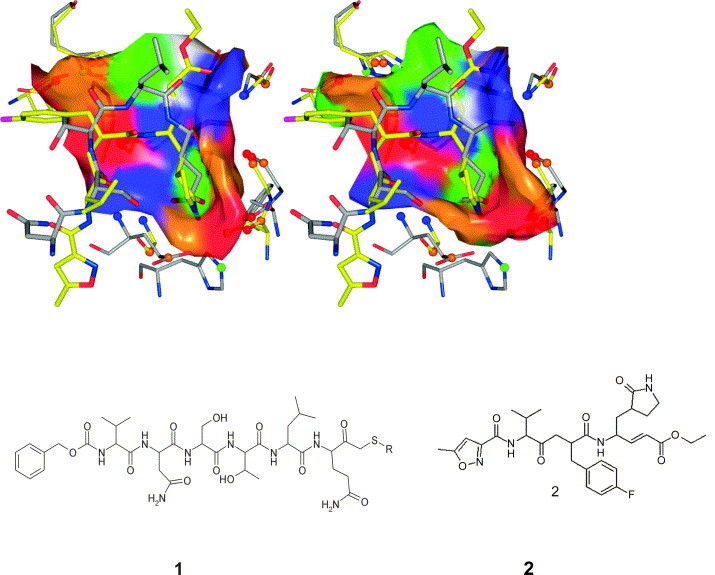
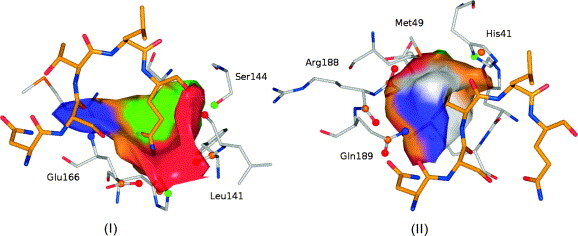
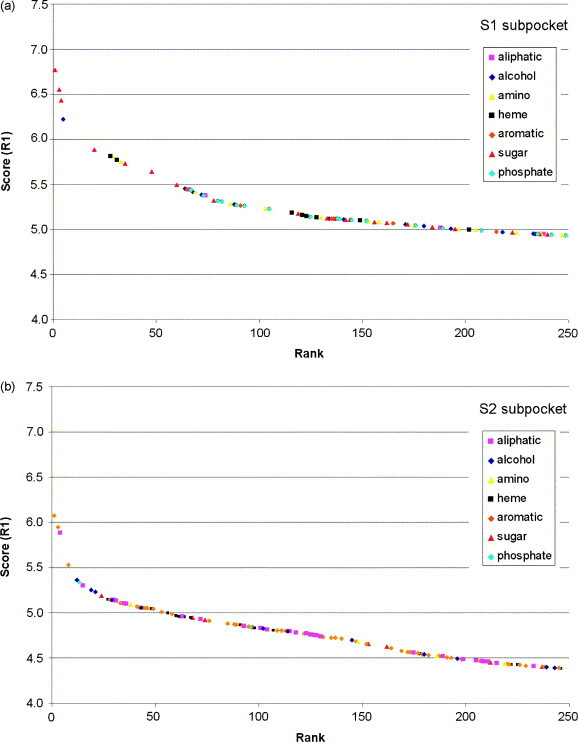
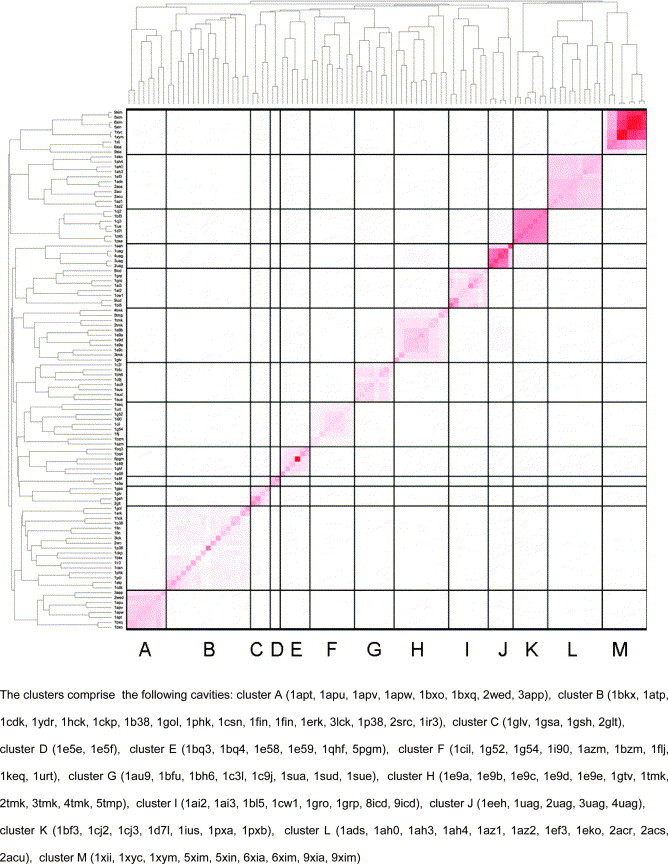
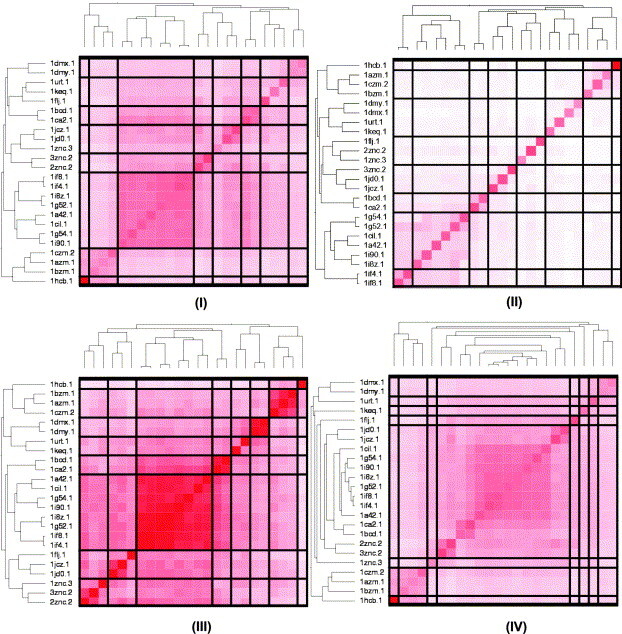
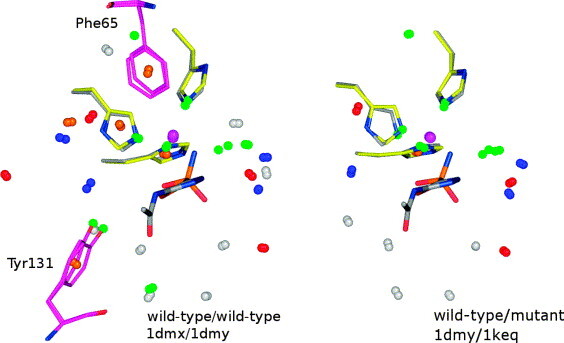
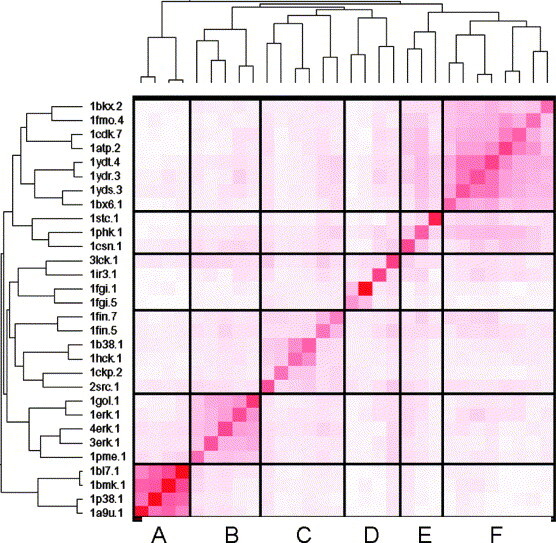
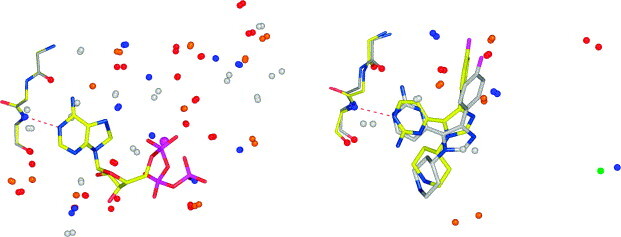
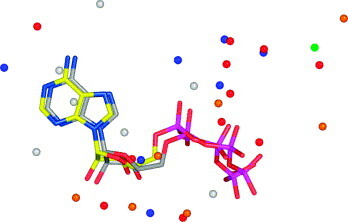
In this contribution, the classification of protein binding sites using the physicochemical properties exposed to their pockets is presented. We recently introduced Cavbase, a method for describing and comparing protein binding pockets on the basis of the geometrical and physicochemical properties of their active sites. Here, we present algorithmic and methodological enhancements in the Cavbase property description and in the cavity comparison step. We give examples of the Cavbase similarity analysis detecting pronounced similarities in the binding sites of proteins unrelated in sequence. A similarity search using SARS M(pro) protease subpockets as queries retrieved ligands and ligand fragments accommodated in a physicochemical environment similar to that of the query. This allowed the characterization of the protease recognition pockets and the identification of molecular building blocks that can be incorporated into novel antiviral compounds. A cluster analysis procedure for the functional classification of binding pockets was implemented and calibrated using a diverse set of enzyme binding sites. Two relevant protein families, the alpha-carbonic anhydrases and the protein kinases, are used to demonstrate the scope of our cluster approach. We propose a relevant classification of both protein families, on the basis of the binding motifs in their active sites. The classification provides a new perspective on functional properties across a protein family and is able to highlight features important for potency and selectivity. Furthermore, this information can be used to identify possible cross-reactivities among proteins due to similarities in their binding sites.
Figures

References
-
- Martin A.C., Orengo C.A., Hutchinson E.G., Jones S., Karmirantzou M., Laskowski R.A., et al. Protein folds and functions. Structure. 1998;6:875–884. - PubMed
-
- Orengo C.A., Sillitoe I., Reeves G., Pearl F.M. Review: what can structural classifications reveal about protein evolution? J. Struct. Biol. 2001;134:145–165. - PubMed
-
- Nagano N., Orengo C.A., Thornton J.M. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 2002;321:741–765. - PubMed
-
- Anantharaman V., Aravind L., Koonin E.V. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr. Opin. Chem. Biol. 2003;7:12–20. - PubMed
-
- Weber A., Casini A., Heine A., Kuhn D., Supuran C.T., Scozzafava A., Klebe G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. J. Med. Chem. 2004;47:550–557. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
