

Inferring loss-of-heterozygosity from unpaired tumors using high-density oligonucleotide SNP arrays
- PMID: 16699594
- PMCID: PMC1458964
- DOI: 10.1371/journal.pcbi.0020041
Inferring loss-of-heterozygosity from unpaired tumors using high-density oligonucleotide SNP arrays
Abstract
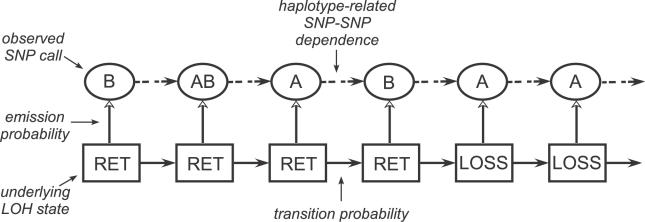
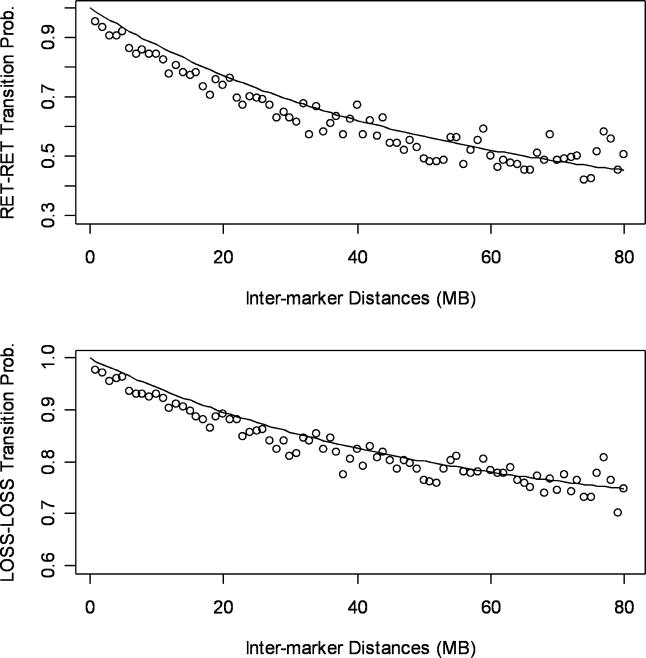
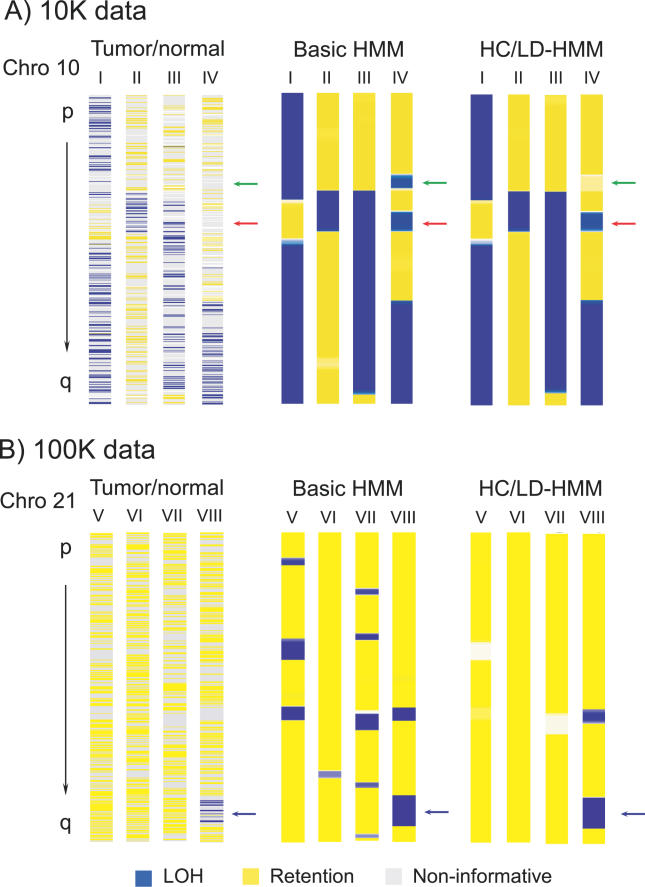
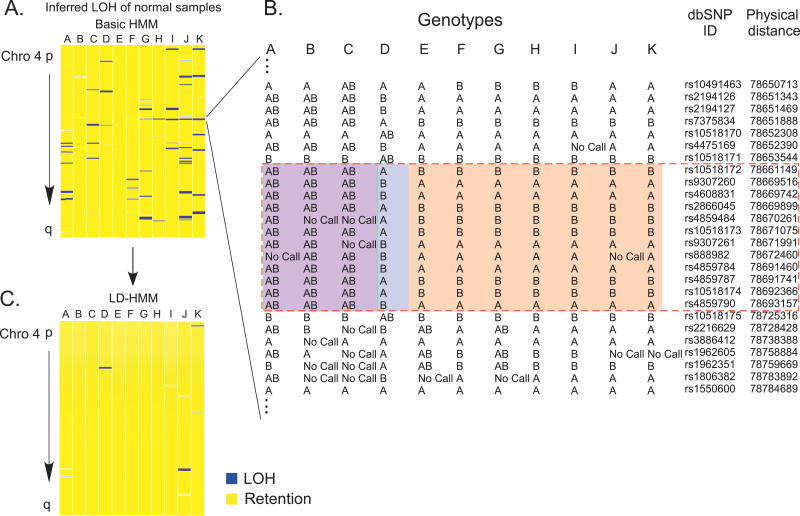
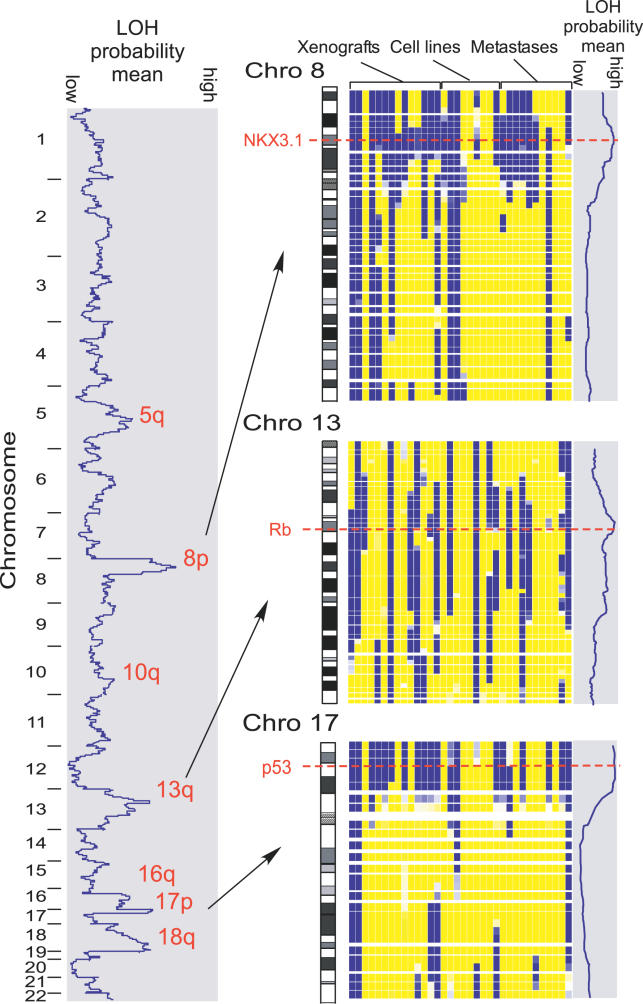
Loss of heterozygosity (LOH) of chromosomal regions bearing tumor suppressors is a key event in the evolution of epithelial and mesenchymal tumors. Identification of these regions usually relies on genotyping tumor and counterpart normal DNA and noting regions where heterozygous alleles in the normal DNA become homozygous in the tumor. However, paired normal samples for tumors and cell lines are often not available. With the advent of oligonucleotide arrays that simultaneously assay thousands of single-nucleotide polymorphism (SNP) markers, genotyping can now be done at high enough resolution to allow identification of LOH events by the absence of heterozygous loci, without comparison to normal controls. Here we describe a hidden Markov model-based method to identify LOH from unpaired tumor samples, taking into account SNP intermarker distances, SNP-specific heterozygosity rates, and the haplotype structure of the human genome. When we applied the method to data genotyped on 100 K arrays, we correctly identified 99% of SNP markers as either retention or loss. We also correctly identified 81% of the regions of LOH, including 98% of regions greater than 3 megabases. By integrating copy number analysis into the method, we were able to distinguish LOH from allelic imbalance. Application of this method to data from a set of prostate samples without paired normals identified known regions of prevalent LOH. We have developed a method for analyzing high-density oligonucleotide SNP array data to accurately identify of regions of LOH and retention in tumors without the need for paired normal samples.
Conflict of interest statement
Figures

References
-
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. - PubMed
-
- McEvoy CR, Morley AA, Firgaira FA. Evidence for whole chromosome 6 loss and duplication of the remaining chromosome in acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2003;37:321–325. - PubMed
-
- Girard L, Zochbauer-Muller S, Virmani AK, Gazdar AF, Minna JD. Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res. 2000;60:4894–4906. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
