

Comprehensive mutation identification in an evolved bacterial cooperator and its cheating ancestor
- PMID: 16707573
- PMCID: PMC1472437
- DOI: 10.1073/pnas.0510740103
Comprehensive mutation identification in an evolved bacterial cooperator and its cheating ancestor
Abstract
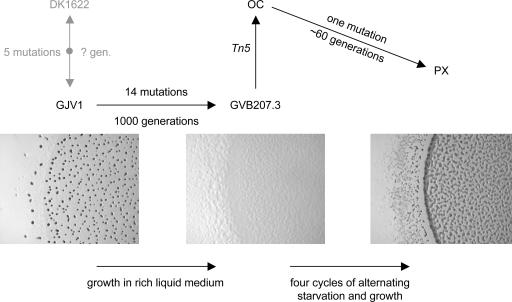
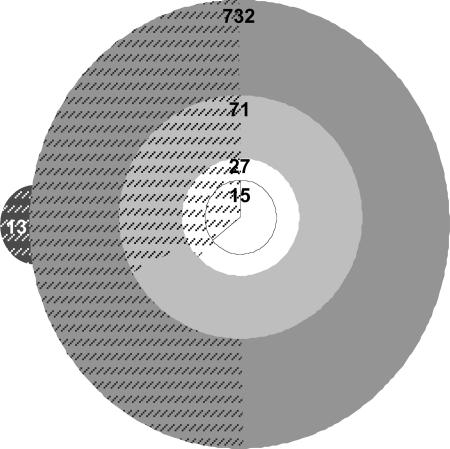
Precise characterization of the mutation histories of evolutionary lineages is crucial for understanding the evolutionary process, yet mutation identification has been constrained by traditional techniques. We sought to identify all accumulated mutations in an experimentally evolved lineage of the cooperative bacterium Myxococcus xanthus, which constructs fruiting bodies by a process of social multicellular development in response to starvation. This lineage had undergone two major transitions in social phenotype: from an ancestral cooperator to a socially defective cheater, and from the cheater to a competitively dominant cooperator that re-evolved social and developmental proficiency. The 9.14-Mb genome of the evolved, dominant cooperator (strain "PX") was sequenced to approximately 19-fold coverage by using recent "sequencing-by-synthesis" technology and partially sequenced (approximately 45%) by using capillary technology. The resulting data revealed 15 single-nucleotide mutations relative to the laboratory ancestor of PX after the two phases of experimental evolution but no evidence of duplications, transpositions, or multiple-base deletions. No mutations were identified by capillary sequencing beyond those found by pyrosequencing, resulting in a high probability that all mutations were discovered. Seven errors in the reference strain previously sequenced by the Sanger approach were revealed, as were five mutational differences between two distinct laboratory stocks of the reference strain. A single mutation responsible for the restoration of development in strain PX was identified, whereas 14 mutations occurred during the prior phase of experimental evolution. These results provide insight into the genetic basis of two large adaptive transitions in a social bacterium.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
