

Refining multiple sequence alignments with conserved core regions
- PMID: 16707662
- PMCID: PMC1463900
- DOI: 10.1093/nar/gkl274
Refining multiple sequence alignments with conserved core regions
Abstract
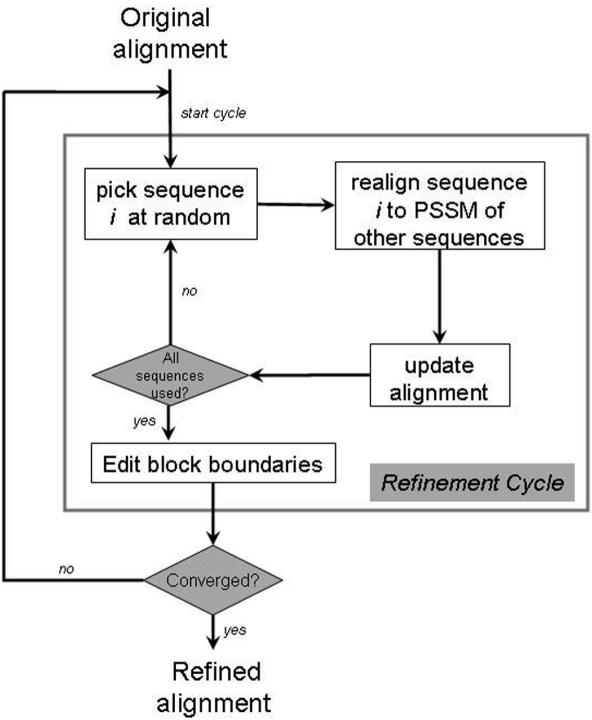
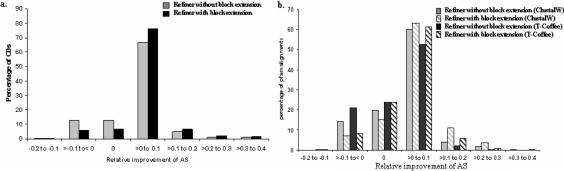
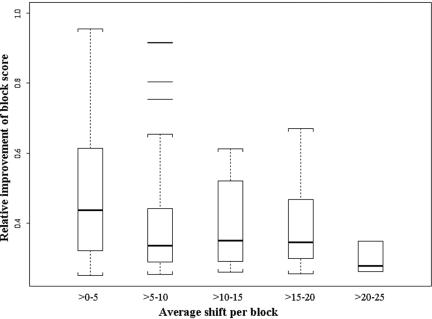
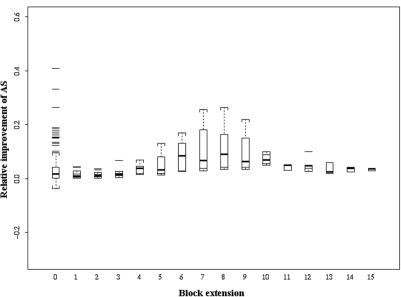
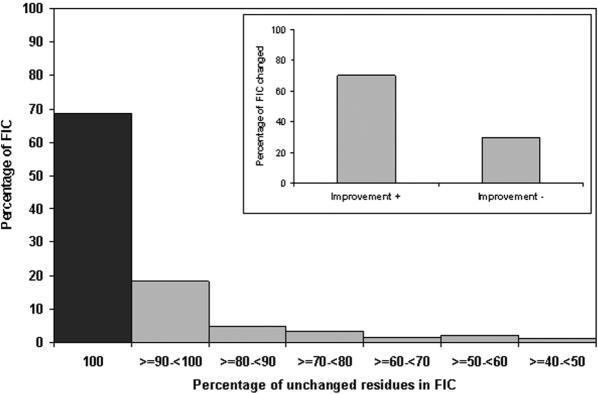
Accurate multiple sequence alignments of proteins are very important to several areas of computational biology and provide an understanding of phylogenetic history of domain families, their identification and classification. This article presents a new algorithm, REFINER, that refines a multiple sequence alignment by iterative realignment of its individual sequences with the predetermined conserved core (block) model of a protein family. Realignment of each sequence can correct misalignments between a given sequence and the rest of the profile and at the same time preserves the family's overall block model. Large-scale benchmarking studies showed a noticeable improvement of alignment after refinement. This can be inferred from the increased alignment score and enhanced sensitivity for database searching using the sequence profiles derived from refined alignments compared with the original alignments. A standalone version of the program is available by ftp distribution (ftp://ftp.ncbi.nih.gov/pub/REFINER) and will be incorporated into the next release of the Cn3D structure/alignment viewer.
Figures

References
-
- Servant F., Bru C., Carrere S., Courcelle E., Gouzy J., Peyruc D., Kahn D. ProDom: Automated clustering of homologous domains. Brief. Bioinformatics. 2002;3:246–251. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
