

Use of a multi-way method to analyze the amino acid composition of a conserved group of orthologous proteins in prokaryotes
- PMID: 16709240
- PMCID: PMC1489954
- DOI: 10.1186/1471-2105-7-257
Use of a multi-way method to analyze the amino acid composition of a conserved group of orthologous proteins in prokaryotes
Abstract
Background: Amino acids in proteins are not used equally. Some of the differences in the amino acid composition of proteins are between species (mainly due to nucleotide composition and lifestyle) and some are between proteins from the same species (related to protein function, expression or subcellular localization, for example). As several factors contribute to the different amino acid usage in proteins, it is difficult both to analyze these differences and to separate the contributions made by each factor.
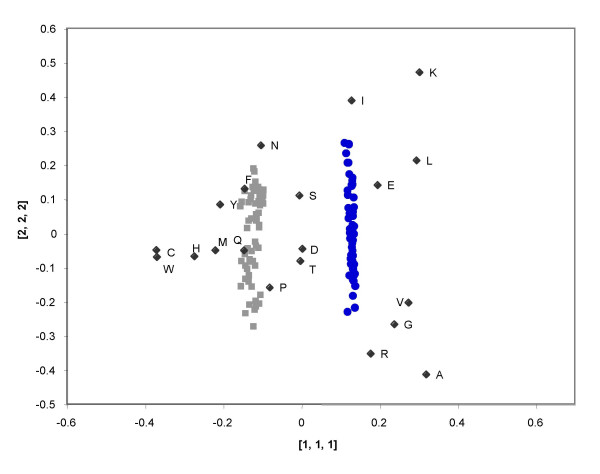
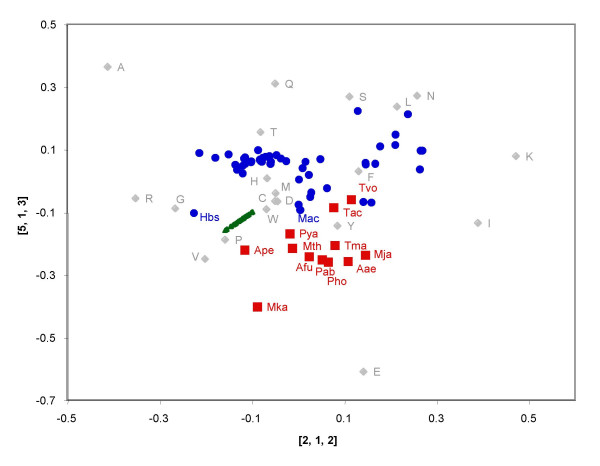
Results: Using a multi-way method called Tucker3, we have analyzed the amino composition of a set of 64 orthologous groups of proteins present in 62 archaea and bacteria. This dataset corresponds to essential proteins such as ribosomal proteins, tRNA synthetases and translational initiation or elongation factors, which are common to all the species analyzed. The Tucker3 model can be used to study the amino acid variability within and between species by taking into consideration the tridimensionality of the data set. We found that the main factor behind the amino acid composition of proteins is independent of the organism or protein function analyzed. This factor must be related to the biochemical characteristics of each amino acid. The difference between the non-ribosomal proteins and the ribosomal proteins (which are rich in arginine and lysine) is the main factor behind the differences in amino acid composition within species, while G+C content and optimal growth temperature are the main factors behind the differences in amino acid usage between species.
Conclusion: We show that a multi-way method is useful for comparing the amino acid composition of several groups of orthologous proteins from the same group of species. This kind of dataset is extremely useful for detecting differences between and within species.
Figures


References
-
- Fujiwara Y, Asogawa M. Prediction of subcellular localizations using amino acid composition and order. Genome Informatics. 2001;12:103–112. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
