

Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study
- PMID: 16709660
- PMCID: PMC1856673
- DOI: 10.1136/gut.2005.087262
Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study
Abstract
Background: Wilson's disease is a rare inborn disease related to copper storage, leading to liver cirrhosis and neuropsychological deterioration. Clinical data on larger cohorts are limited owing to low disease frequency.
Objective and methods: We performed a retrospective analysis of 163 patients with Wilson's disease, examined at the University of Heidelberg, Heidelberg, Germany, to determine clinical presentation, diagnostic course and long-term outcome.
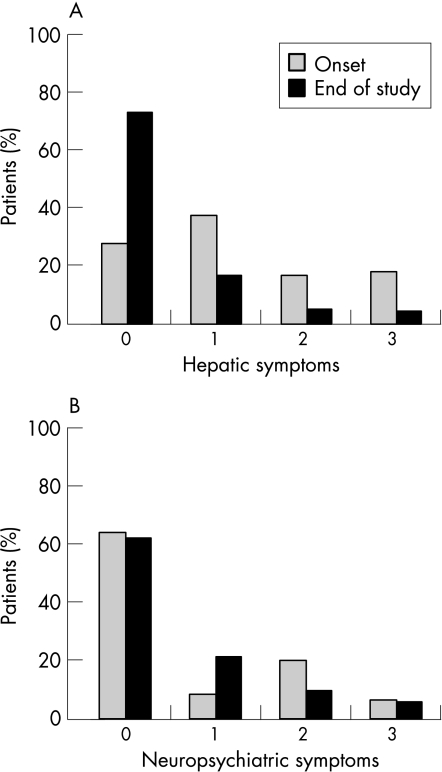
Results: Diagnostic criteria for non-caeruloplasmin-bound serum copper, serum caeruloplasmin, 24-h urinary copper excretion, liver copper content, presence of Kayser-Fleischer rings and histological signs of chronic liver damage were reached in 86.6%, 88.2%, 87.1%, 92.7%, 66.3% and 73% of patients, respectively. By analysis of the coding region of ATP7B (except exons 2, 3 and 21), disease-causing mutations were detected in 57% and 29% of patients with Wilson's disease on both chromosomes and on one chromosome, respectively. No mutations were detected in 15% of patients with Wilson's disease. No significant differences were found in clinical parameters or initial presentation between patients grouped according to their mutations. The patients with neurological symptoms were significantly older at the onset of symptoms than patients with hepatitic symptoms (20.2 v 15.5 years of age, p<0.05), and the neurological symptoms were associated with a significantly longer time from onset to diagnosis than hepatic symptoms (44.4 v 14.4 months, p<0.05). After initiating treatment, 76.1% of the patients had a stable or improved course of the disease. Disease progression under treatment was more likely for neuropsychiatric than for hepatic symptoms. Side effects of treatment occurred in 74.4% of patients.
Conclusions: Patients with Wilson's disease having predominantly neuropsychiatric symptoms manifest symptoms later, have a longer time delay from onset of symptoms until definitive diagnosis and have a poorer outcome than patients with hepatic symptoms.
Conflict of interest statement
Competing interests: None.


Comment in
-
Limitations of non-ceruloplasmin-bound copper in routine clinical practice.Gut. 2007 Jan;56(1):154. Gut. 2007. PMID: 17172594 Free PMC article. No abstract available.
References
-
- Scheinberg I H. Wilson's disease. J Rheumatol Suppl 1981790–93. - PubMed
-
- Bull P C, Thomas G R, Rommens J M.et al The Wilson disease gene is a putative copper transporting P‐type ATPase similar to the Menkes gene. Nat Genet 19935327–337. - PubMed
-
- Tanzi R E, Petrukhin K, Chernov I.et al The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 19935344–350. - PubMed
-
- Stremmel W, Meyerrose K W, Niederau C.et al Wilson disease: clinical presentation, treatment, and survival. Ann Intern Med 1991115720–726. - PubMed
-
- Gitlin J D. Wilson disease. Gastroenterology 20031251868–1877. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical