

Statistics of knots, geometry of conformations, and evolution of proteins
- PMID: 16710448
- PMCID: PMC1463020
- DOI: 10.1371/journal.pcbi.0020045
Statistics of knots, geometry of conformations, and evolution of proteins
Abstract
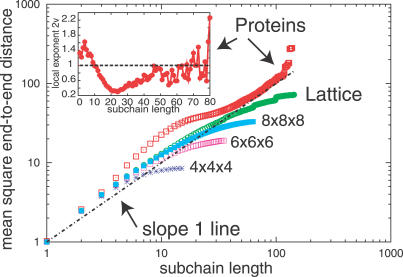
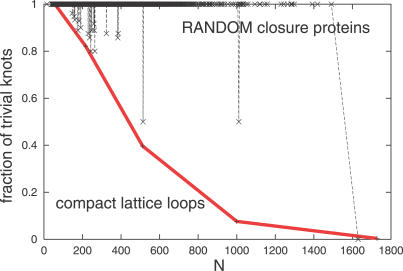
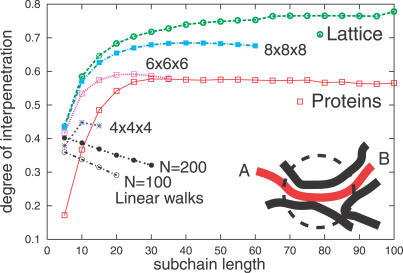
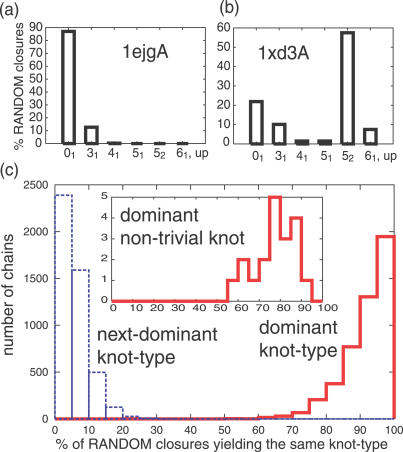
Like shoelaces, the backbones of proteins may get entangled and form knots. However, only a few knots in native proteins have been identified so far. To more quantitatively assess the rarity of knots in proteins, we make an explicit comparison between the knotting probabilities in native proteins and in random compact loops. We identify knots in proteins statistically, applying the mathematics of knot invariants to the loops obtained by complementing the protein backbone with an ensemble of random closures, and assigning a certain knot type to a given protein if and only if this knot dominates the closure statistics (which tells us that the knot is determined by the protein and not by a particular method of closure). We also examine the local fractal or geometrical properties of proteins via computational measurements of the end-to-end distance and the degree of interpenetration of its subchains. Although we did identify some rather complex knots, we show that native conformations of proteins have statistically fewer knots than random compact loops, and that the local geometrical properties, such as the crumpled character of the conformations at a certain range of scales, are consistent with the rarity of knots. From these, we may conclude that the known "protein universe" (set of native conformations) avoids knots. However, the precise reason for this is unknown--for instance, if knots were removed by evolution due to their unfavorable effect on protein folding or function or due to some other unidentified property of protein evolution.
Conflict of interest statement
Figures
References
-
- Mansfield ML. Are there knots in proteins? Nat Struct Biol. 1994;1:213–214. - PubMed
-
- Mansfield ML. Fit to be tied. Nat Struct Biol. 1997;4:166–167. - PubMed
-
- Taylor WR. A deeply knotted protein and how it might fold. Nature. 2000;406:916–919. - PubMed
-
- Taylor WR, Lin K. A tangled problem. Nature. 2003;421:25. - PubMed
-
- Taylor WR, May ACW, Brown NP, Aszodi A. Protein structure: Geometry, topology and classification. Rep Prog Phys. 2001;64:517–590.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
