

Multiple sequence alignment with user-defined anchor points
- PMID: 16722533
- PMCID: PMC1481597
- DOI: 10.1186/1748-7188-1-6
Multiple sequence alignment with user-defined anchor points
Abstract
Background: Automated software tools for multiple alignment often fail to produce biologically meaningful results. In such situations, expert knowledge can help to improve the quality of alignments.
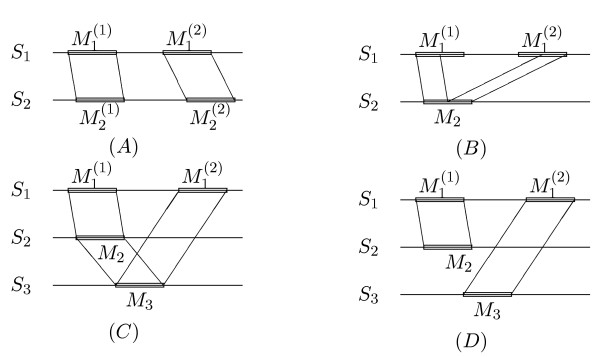
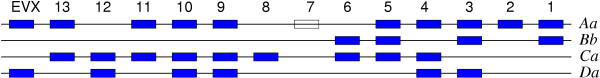
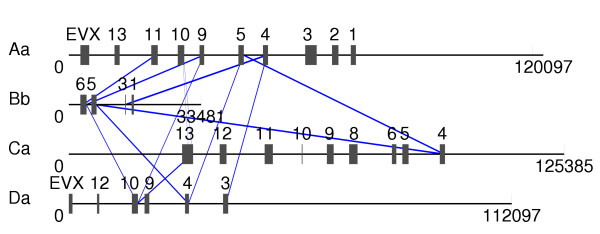
Results: Herein, we describe a semi-automatic version of the alignment program DIALIGN that can take pre-defined constraints into account. It is possible for the user to specify parts of the sequences that are assumed to be homologous and should therefore be aligned to each other. Our software program can use these sites as anchor points by creating a multiple alignment respecting these constraints. This way, our alignment method can produce alignments that are biologically more meaningful than alignments produced by fully automated procedures. As a demonstration of how our method works, we apply our approach to genomic sequences around the Hox gene cluster and to a set of DNA-binding proteins. As a by-product, we obtain insights about the performance of the greedy algorithm that our program uses for multiple alignment and about the underlying objective function. This information will be useful for the further development of DIALIGN. The described alignment approach has been integrated into the TRACKER software system.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
