

Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome
- PMID: 16722536
- PMCID: PMC1475558
- DOI: 10.1186/1750-1172-1-13
Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome
Abstract
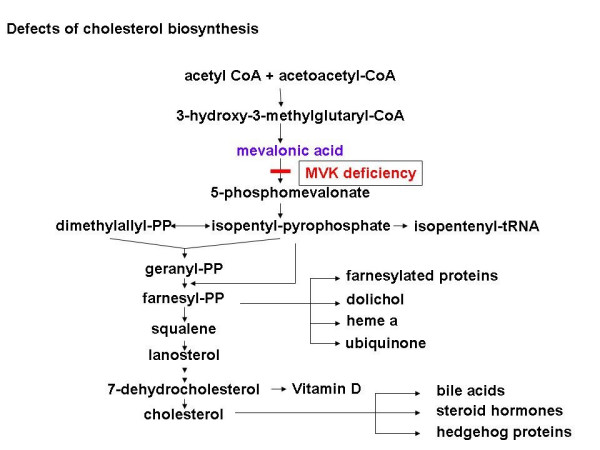
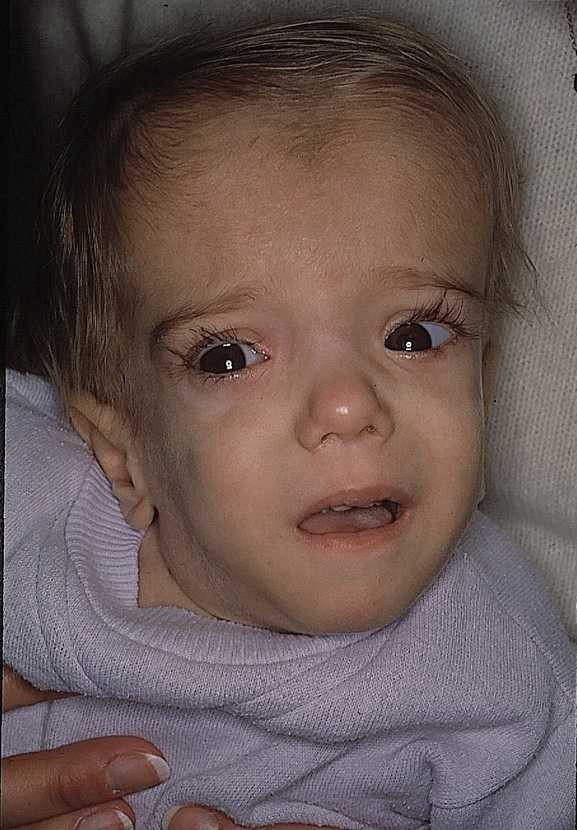
Mevalonic aciduria (MVA) and hyperimmunoglobulinemia D syndrome (HIDS) represent the two ends of a clinical spectrum of disease caused by deficiency of mevalonate kinase (MVK), the first committed enzyme of cholesterol biosynthesis. At least 30 patients with MVA and 180 patients with HIDS have been reported worldwide. MVA is characterized by psychomotor retardation, failure to thrive, progressive cerebellar ataxia, dysmorphic features, progressive visual impairment and recurrent febrile crises. The febrile episodes are commonly accompanied by hepatosplenomegaly, lymphadenopathy, abdominal symptoms, arthralgia and skin rashes. Life expectancy is often compromised. In HIDS, only febrile attacks are present, but a subgroup of patients may also develop neurological abnormalities of varying degree such as mental retardation, ataxia, ocular symptoms and epilepsy. A reduced activity of MVK and pathogenic mutations in the MVK gene have been demonstrated as the common genetic basis in both disorders. In MVA, the diagnosis is established by detection of highly elevated levels of mevalonic acid excreted in urine. Increased levels of immunoglobulin D (IgD) and, in most patients of immunoglobulin A (IgA), in combination with enhanced excretion of mevalonic acid provide strong evidence for HIDS. The diagnosis is confirmed by low activity of mevalonate kinase or by demonstration of disease-causing mutations. Genetic counseling should be offered to families at risk. There is no established successful treatment for MVA. Simvastatin, an inhibitor of HMG-CoA reductase, and anakinra have been shown to have beneficial effect in HIDS.
Figures
References
-
- Hoffmann G, Gibson KM, Brandt IK, Bader PI, Wappner RS, Sweetman L. Mevalonic aciduria – an inborn error of cholesterol and nonsterol isoprene biosynthesis. N Engl J Med. 1986;314:1610–1614. - PubMed
-
- Simon A, Kremer HP, Wevers RA, Scheffer H, De Jong JG, Van Der Meer JW, Drenth JP. Mevalonate kinase deficiency – Evidence for a phenotypic continuum. Neurology. 2004;62:994–997. - PubMed
-
- Hoffmann GF, Charpentier C, Mayatepek E, Mancini J, Leichsenring M, Gibson KM, Divry P, Hrebicek M, Lehnert W, Sartor K, et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics. 1993;91:915–921. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
