

ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context
- PMID: 16723010
- PMCID: PMC1810318
- DOI: 10.1186/1471-2105-7-S1-S7
ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context
Abstract
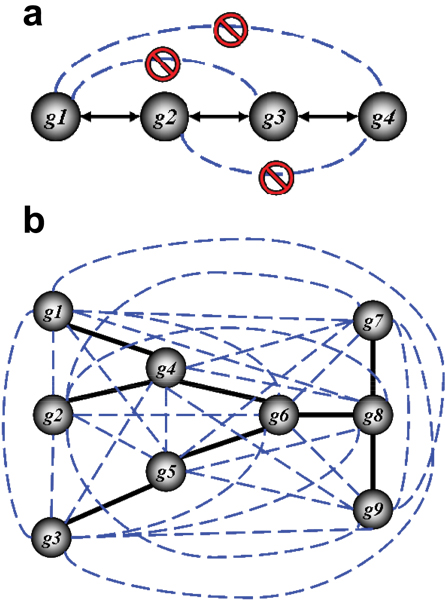
Background: Elucidating gene regulatory networks is crucial for understanding normal cell physiology and complex pathologic phenotypes. Existing computational methods for the genome-wide "reverse engineering" of such networks have been successful only for lower eukaryotes with simple genomes. Here we present ARACNE, a novel algorithm, using microarray expression profiles, specifically designed to scale up to the complexity of regulatory networks in mammalian cells, yet general enough to address a wider range of network deconvolution problems. This method uses an information theoretic approach to eliminate the majority of indirect interactions inferred by co-expression methods.
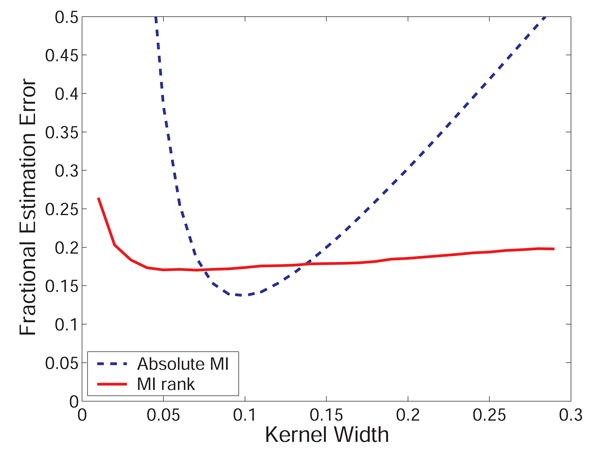
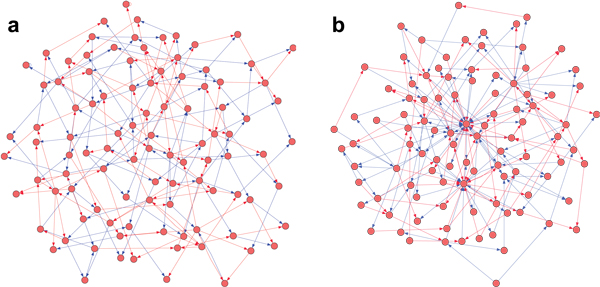
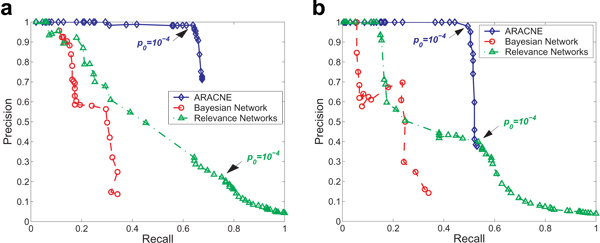
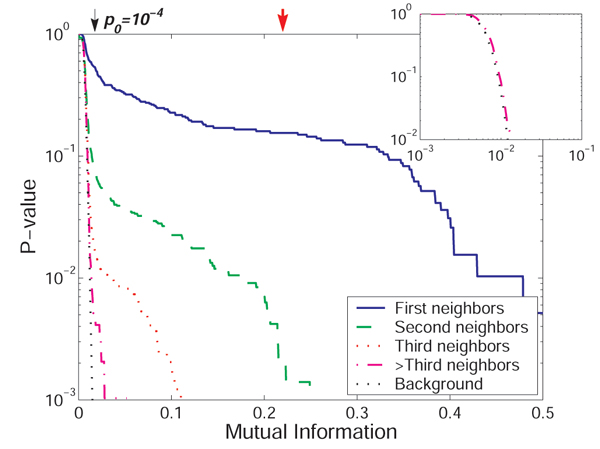
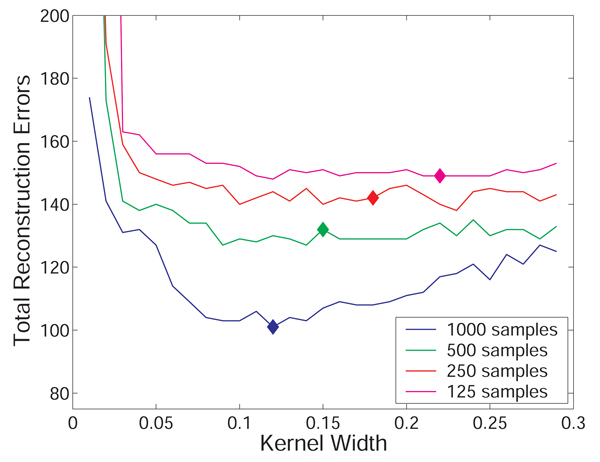
Results: We prove that ARACNE reconstructs the network exactly (asymptotically) if the effect of loops in the network topology is negligible, and we show that the algorithm works well in practice, even in the presence of numerous loops and complex topologies. We assess ARACNE's ability to reconstruct transcriptional regulatory networks using both a realistic synthetic dataset and a microarray dataset from human B cells. On synthetic datasets ARACNE achieves very low error rates and outperforms established methods, such as Relevance Networks and Bayesian Networks. Application to the deconvolution of genetic networks in human B cells demonstrates ARACNE's ability to infer validated transcriptional targets of the cMYC proto-oncogene. We also study the effects of misestimation of mutual information on network reconstruction, and show that algorithms based on mutual information ranking are more resilient to estimation errors.
Conclusion: ARACNE shows promise in identifying direct transcriptional interactions in mammalian cellular networks, a problem that has challenged existing reverse engineering algorithms. This approach should enhance our ability to use microarray data to elucidate functional mechanisms that underlie cellular processes and to identify molecular targets of pharmacological compounds in mammalian cellular networks.
Figures
References
-
- Ma S-K. Statistical mechanics. Singapore: World Scientific; 1985.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
