

A uniform proteomics MS/MS analysis platform utilizing open XML file formats
- PMID: 16729052
- PMCID: PMC1681455
- DOI: 10.1038/msb4100024
A uniform proteomics MS/MS analysis platform utilizing open XML file formats
Abstract
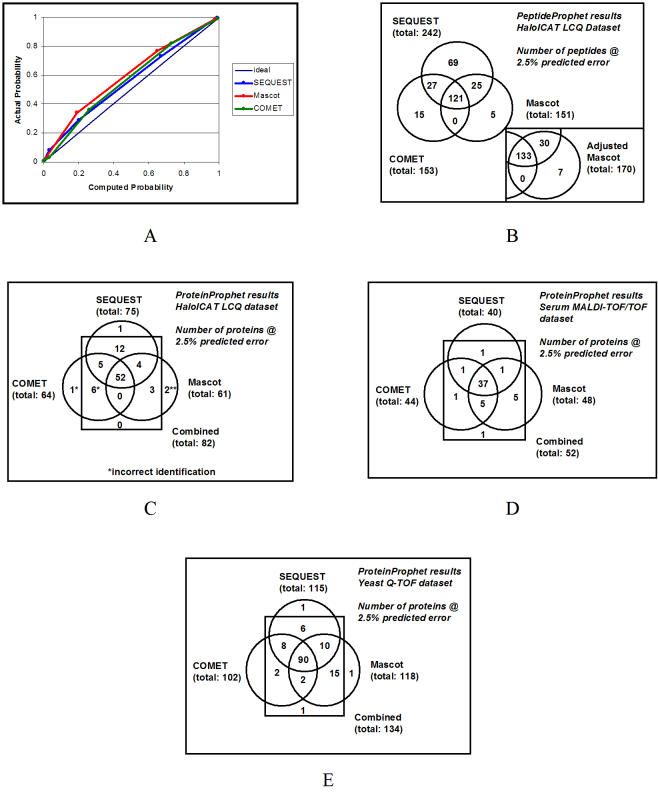
The analysis of tandem mass (MS/MS) data to identify and quantify proteins is hampered by the heterogeneity of file formats at the raw spectral data, peptide identification, and protein identification levels. Different mass spectrometers output their raw spectral data in a variety of proprietary formats, and alternative methods that assign peptides to MS/MS spectra and infer protein identifications from those peptide assignments each write their results in different formats. Here we describe an MS/MS analysis platform, the Trans-Proteomic Pipeline, which makes use of open XML file formats for storage of data at the raw spectral data, peptide, and protein levels. This platform enables uniform analysis and exchange of MS/MS data generated from a variety of different instruments, and assigned peptides using a variety of different database search programs. We demonstrate this by applying the pipeline to data sets generated by ThermoFinnigan LCQ, ABI 4700 MALDI-TOF/TOF, and Waters Q-TOF instruments, and searched in turn using SEQUEST, Mascot, and COMET.
Figures

References
-
- Chen SC, Deutsch EW, Yi EC, Li X-J, Goodlett DR, Aebersold R Improving mass and liquid chromatography based identification of proteins using Bayesian scoring. (manuscript in preparation) - PubMed
-
- Craig R, Beavis RC (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 12: 1466–1467 - PubMed
-
- Eng J, Martin DB, Aebersold R (2005) Tandem mass spectrometry database searching. In Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics, Dunn M, Jorde L, Little P, Subramaniam S (eds). John Wiley & Sons, Ltd, ISB 0470849746
-
- Field HI, Fenyo D, Beavis RC (2002) RADARS, a bioinformatics solution that automates proteome mass spectral analysis, optimises protein identification, and archives data in a relational database. Proteomics 2: 36–47 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
