

Epithelial origin of myofibroblasts during fibrosis in the lung
- PMID: 16738204
- PMCID: PMC2658689
- DOI: 10.1513/pats.200601-004TK
Epithelial origin of myofibroblasts during fibrosis in the lung
Abstract
An understanding of the mechanisms underlying pulmonary fibrosis remains elusive. Once believed to result primarily from chronic inflammation, it is now clear that inflammation and chronic fibrosis, especially in diseases such as idiopathic pulmonary fibrosis/usual interstitial pneumonia, are often dissociated, and that inflammation is neither necessary nor sufficient to induce fibrosis. The origin of the primary effector cell of fibrosis in the lung, the myofibroblast, is not clearly established. Three potential sources have been hypothesized. Although conversion of resident fibroblasts and differentiation of circulating bone marrow-derived progenitors likely play a role, the possible contribution of alveolar epithelial cells (AECs), through a process termed "epithelial-mesenchymal transition" (EMT), has only recently received consideration. A process by which epithelial cells lose cell-cell attachment, polarity and epithelial-specific markers, undergo cytoskeletal remodeling, and gain a mesenchymal phenotype, EMT plays a prominent role in fibrogenesis in adult tissues such as the kidney. This review summarizes the evidence supporting a central role for EMT in the pathogenesis of lung fibrosis, the potential for EMT in AECs in vitro and in vivo and role of transforming growth factor-beta1 in this process, and the implications of epithelium-driven fibrosis on future research and treatment. Potential pathways involved in EMT are also discussed. It is hoped that a major shift in current paradigms regarding the genesis of pulmonary fibrosis and dissection of the relevant pathways may allow development of targeted interventions that could potentially reverse the process and ameliorate the debilitating effects of abnormal repair and progressive fibrosis.
Figures
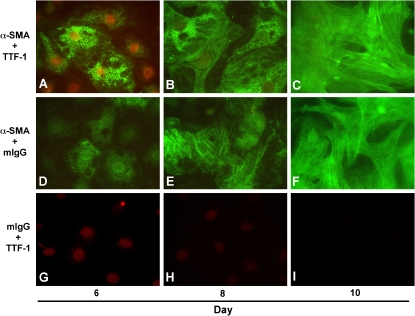
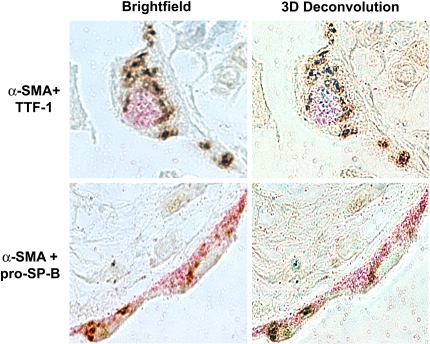
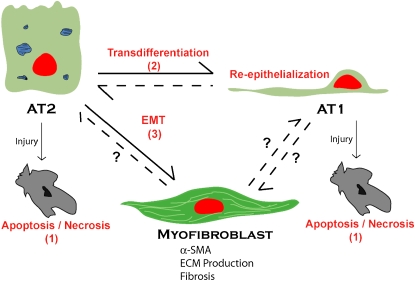
References
-
- Du Bois RM, Wells AU. Cryptogenic fibrosing alveolitis/idiopathic pulmonary fibrosis. Eur Respir J Suppl 2001;32:43s–55s. - PubMed
-
- Munger JS, Huang X, Kawakatsu H, Griffiths MJD, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin αvβ6 binds and activates latent TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999;96:319–328. - PubMed
-
- Pardo A, Selman M. Idiopathic pulmonary fibrosis: new insights in its pathogenesis. Int J Biochem Cell Biol 2002;34:1534–1538. - PubMed
-
- Selman M, Pardo A. The epithelial/fibroblastic pathway in the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2003;29:S93–S96. - PubMed
-
- King TE Jr, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JA Jr, Flint A, Thurlbeck W, Cherniack RM. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001;164:1025–1032. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical