

Systems-level analyses identify extensive coupling among gene expression machines
- PMID: 16738550
- PMCID: PMC1681477
- DOI: 10.1038/msb4100045
Systems-level analyses identify extensive coupling among gene expression machines
Abstract
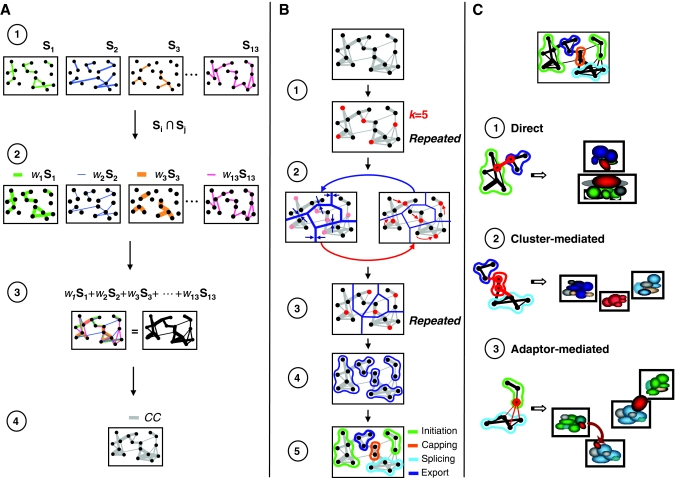
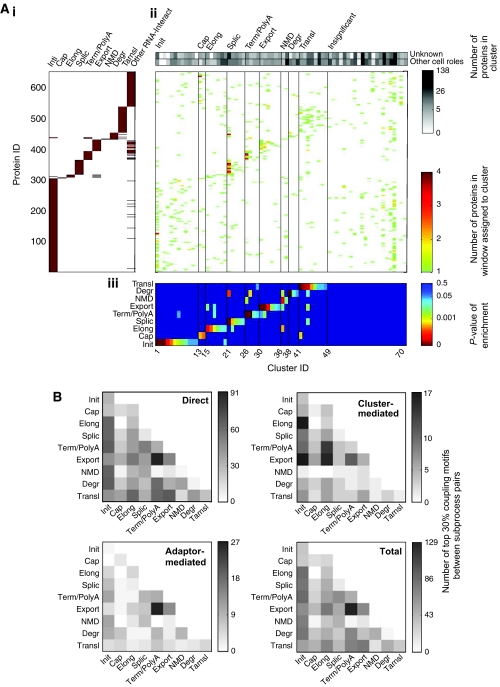
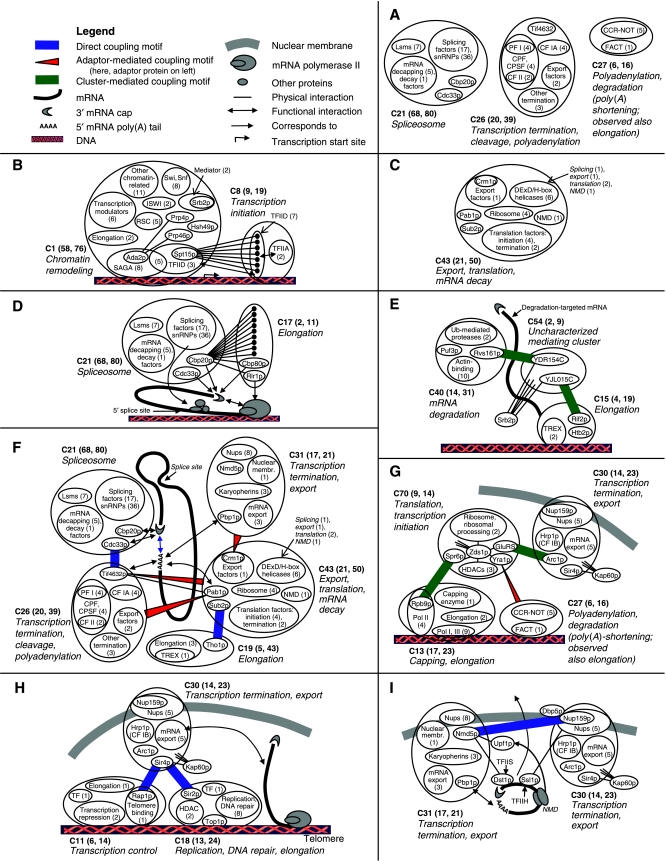
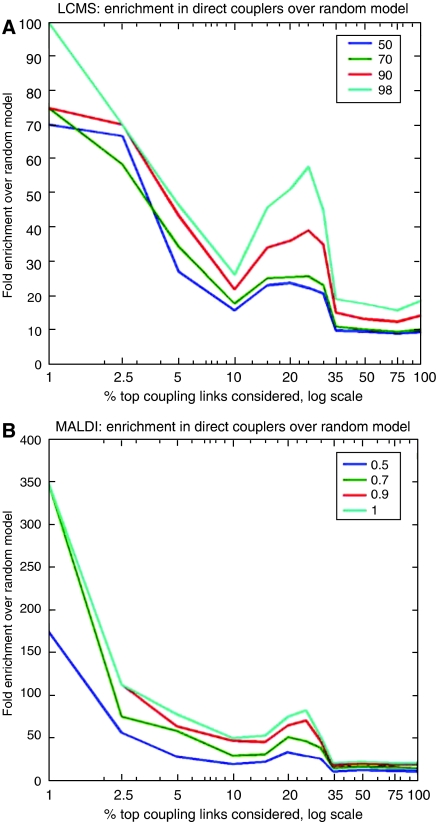
Here, we develop computational methods to assess and consolidate large, diverse protein interaction data sets, with the objective of identifying proteins involved in the coupling of multicomponent complexes within the yeast gene expression pathway. From among approximately 43 000 total interactions and 2100 proteins, our methods identify known structural complexes, such as the spliceosome and SAGA, and functional modules, such as the DEAD-box helicases, within the interaction network of proteins involved in gene expression. Our process identifies and ranks instances of three distinct, biologically motivated motifs, or patterns of coupling among distinct machineries involved in different subprocesses of gene expression. Our results confirm known coupling among transcription, RNA processing, and export, and predict further coupling with translation and nonsense-mediated decay. We systematically corroborate our analysis with two independent, comprehensive experimental data sets. The methods presented here may be generalized to other biological processes and organisms to generate principled, systems-level network models that provide experimentally testable hypotheses for coupling among biological machines.
Figures
References
-
- Aguilera A (2005) Cotranscriptional mRNP assembly: from the DNA to the nuclear pore. Curr Opin Cell Biol 17: 242–250 - PubMed
-
- Ares M Jr, Proudfoot NJ (2005) The spanish connection: transcription and mRNA processing get even closer. Cell 120: 163–166 - PubMed
-
- Auty R, Steen H, Myers LC, Persinger J, Bartholomew B, Gygi SP, Buratowski S (2004) Purification of active TFIID from Saccharomyces cerevisiae. Extensive promoter contacts and co-activator function. J Biol Chem 279: 49973–49981 - PubMed
-
- Bader GD, Hogue CW (2002) Analyzing yeast protein–protein interaction data obtained from different sources. Nat Biotechnol 20: 991–997 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
