

Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction
- PMID: 16741575
- PMCID: PMC1464895
- DOI: 10.1172/JCI25397
Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction
Abstract
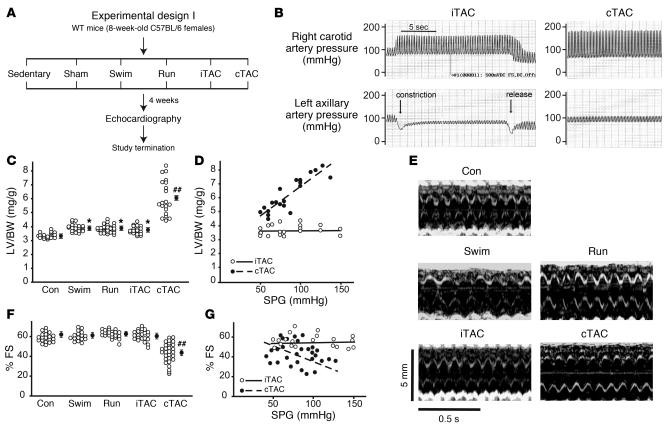
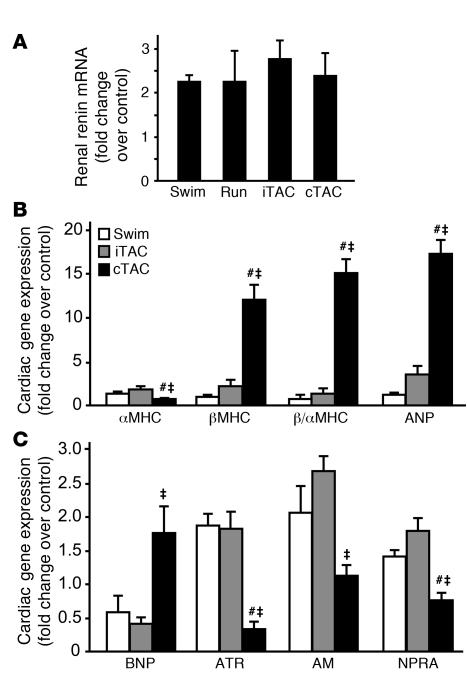
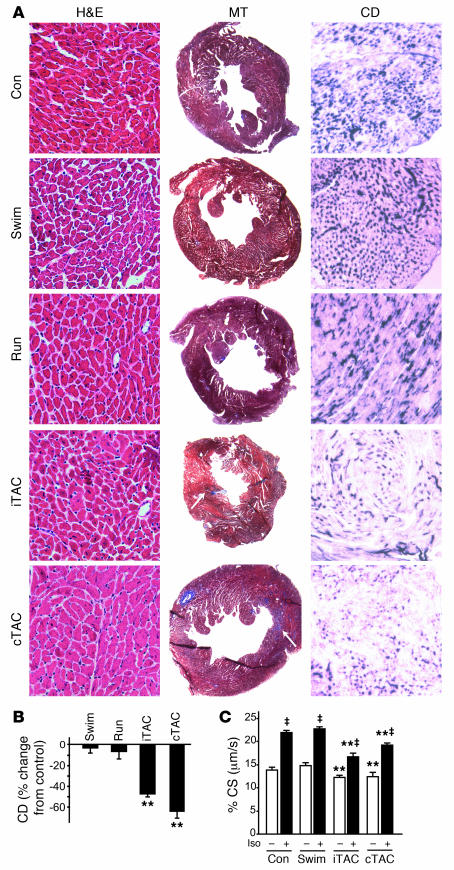
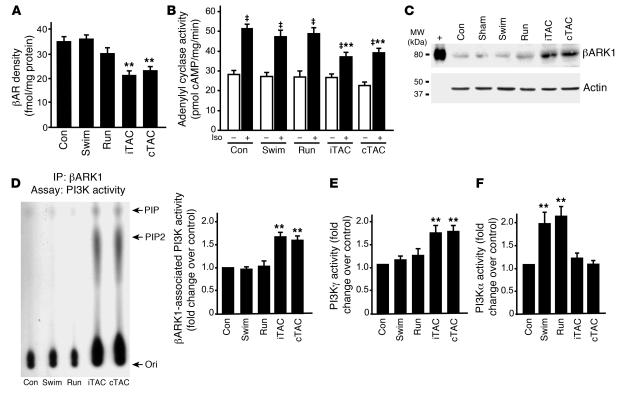
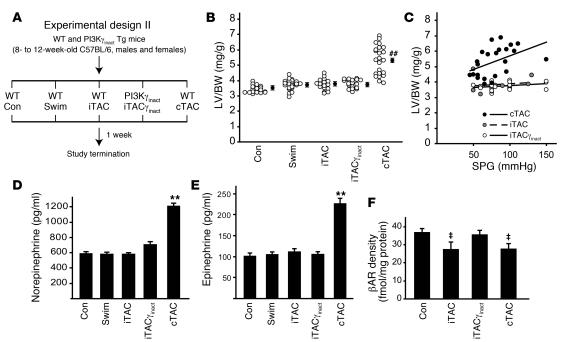
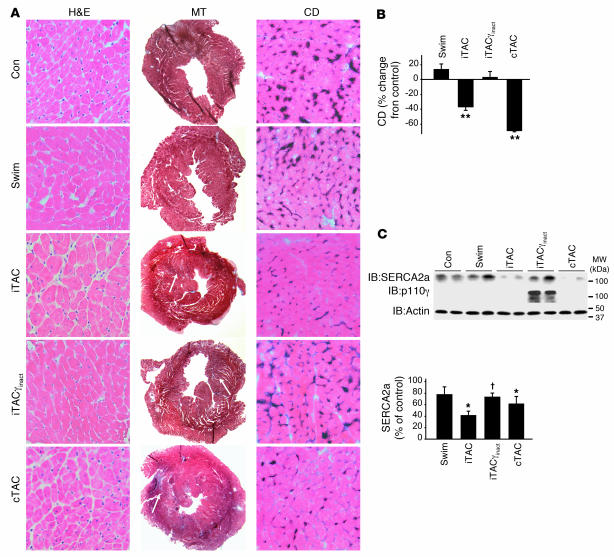
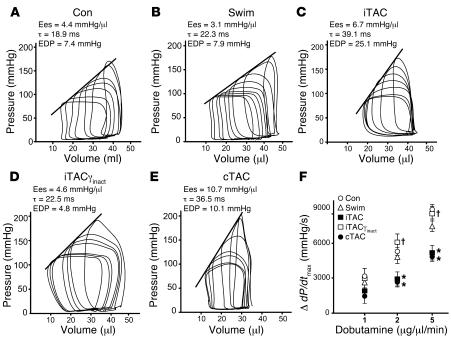
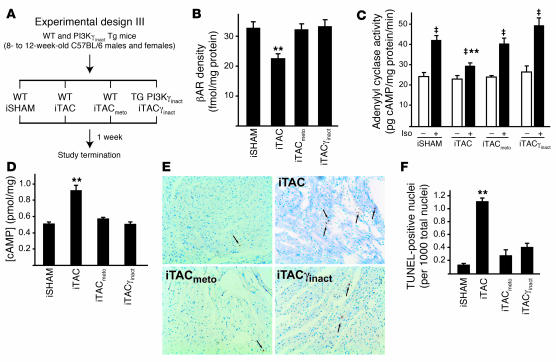
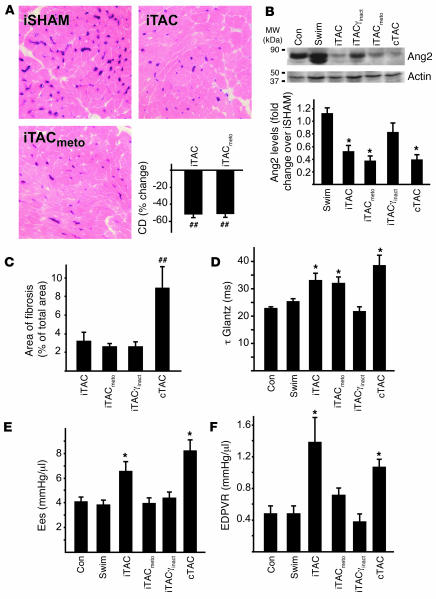
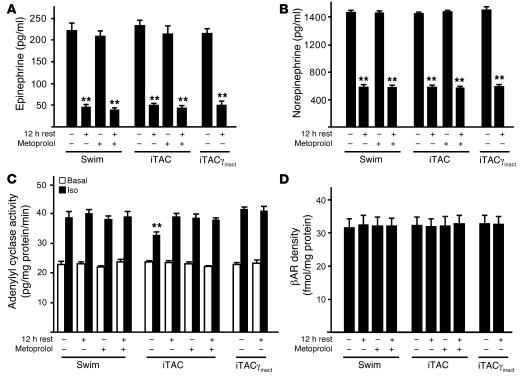
For over a century, there has been intense debate as to the reason why some cardiac stresses are pathological and others are physiological. One long-standing theory is that physiological overloads such as exercise are intermittent, while pathological overloads such as hypertension are chronic. In this study, we hypothesized that the nature of the stress on the heart, rather than its duration, is the key determinant of the maladaptive phenotype. To test this, we applied intermittent pressure overload on the hearts of mice and tested the roles of duration and nature of the stress on the development of cardiac failure. Despite a mild hypertrophic response, preserved systolic function, and a favorable fetal gene expression profile, hearts exposed to intermittent pressure overload displayed pathological features. Importantly, intermittent pressure overload caused diastolic dysfunction, altered beta-adrenergic receptor (betaAR) function, and vascular rarefaction before the development of cardiac hypertrophy, which were largely normalized by preventing the recruitment of PI3K by betaAR kinase 1 to ligand-activated receptors. Thus stress-induced activation of pathogenic signaling pathways, not the duration of stress or the hypertrophic growth per se, is the molecular trigger of cardiac dysfunction.
Figures
Comment in
-
Cardiac hypertrophy: stressing out the heart.J Clin Invest. 2006 Jun;116(6):1467-70. doi: 10.1172/JCI28884. J Clin Invest. 2006. PMID: 16741569 Free PMC article.
References
-
- Meerson F.Z. Compensatory hyperfunction of the heart and cardiac insufficiency. Circ. Res. 1962;10:250–258. - PubMed
-
- Levy D., Garrison R.J., Savage D.D., Kannel W.B., Castelli W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 1990;322:1561–1566. - PubMed
-
- Frey N., Katus H.A., Olson E.N., Hill J.A. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. - PubMed
-
- Esposito G., et al. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92. - PubMed
-
- Yusuf S., et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N. Engl. J. Med. 2000;342:145–153. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
