

Genetic and pharmacological evidence that a retinoic acid cannot be the RXR-activating ligand in mouse epidermis keratinocytes
- PMID: 16751185
- PMCID: PMC1475764
- DOI: 10.1101/gad.368706
Genetic and pharmacological evidence that a retinoic acid cannot be the RXR-activating ligand in mouse epidermis keratinocytes
Abstract
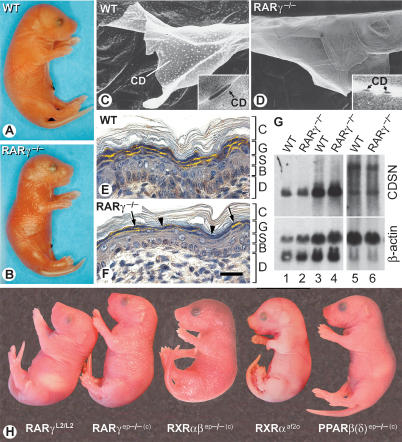
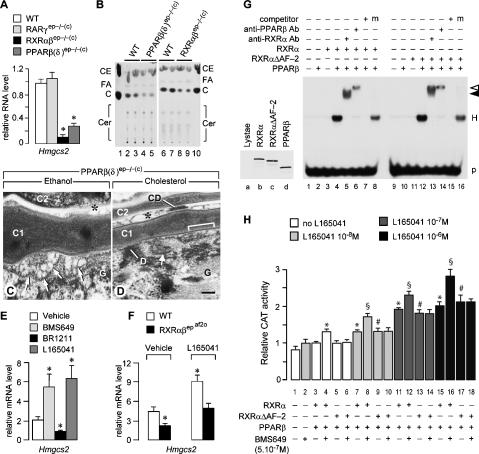
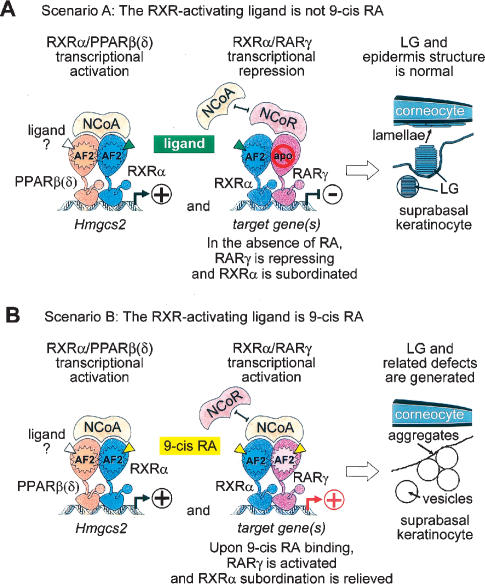
Using genetic and pharmacological approaches, we demonstrate that both RARgamma/RXRalpha heterodimers involved in repression events, as well as PPARbeta(delta)/RXRalpha heterodimers involved in activation events, are cell-autonomously required in suprabasal keratinocytes for the generation of lamellar granules (LG), the organelles instrumental to the formation of the skin permeability barrier. In activating PPARbeta(delta)/RXRalpha heterodimers, RXRalpha is transcriptionally active as its AF-2 activation function is required and can be inhibited by an RXR-selective antagonist. Within repressing RARgamma/RXRalpha heterodimers, induction of the transcriptional activity of RXRalpha is subordinated to the addition of an agonistic ligand for RARgamma. Thus, the ligand that possibly binds and activates RXRalpha heterodimerized with PPARbeta(delta) cannot be a retinoic acid, as it would also bind RARgamma and relieve the RARgamma-mediated repression, thereby yielding abnormal LGs. Our data also demonstrate for the first time that subordination of RXR transcriptional activity to that of its RAR partner plays a crucial role in vivo, because it allows RXRs to act concomitantly, within the same cell, as heterodimerization partners for repression, as well as for activation events in which they are transcriptionally active.
Figures




References
-
- Abu-Abed S., MacLean G., Fraulob V., Chambon P., Petkovich M., Dolle P. Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech. Dev. 2002;110:173–177. - PubMed
-
- Andersen B., Rosenfeld M.G. Intracellular receptors. New wrinkles in retinoids. Nature. 1995;374:118–119. - PubMed
-
- Arnold M.L., Anton-Lamprecht I., Melz-Rothfuss B., Hartschuh W. Ichthyosis congenita type III. Clinical and ultrastructural characteristics and distinction within the heterogeneous ichthyosis congenita group. Arch. Dermatol. Res. 1988;280:268–278. - PubMed
-
- Attar P.S., Wertz P.W., McArthur M., Imakado S., Bickenbach J.R., Roop D.R. Inhibition of retinoid signaling in transgenic mice alters lipid processing and disrupts epidermal barrier function. Mol. Endocrinol. 1997;11:792–800. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases