

Scan of human genome reveals no new Loci under ancient balancing selection
- PMID: 16751668
- PMCID: PMC1569689
- DOI: 10.1534/genetics.106.055715
Scan of human genome reveals no new Loci under ancient balancing selection
Abstract
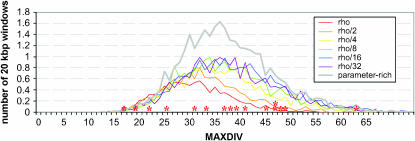
There has been much speculation as to what role balancing selection has played in evolution. In an attempt to identify regions, such as HLA, at which polymorphism has been maintained in the human population for millions of years, we scanned the human genome for regions of high SNP density. We found 16 regions that, outside of HLA and ABO, are the most highly polymorphic regions yet described; however, evidence for balancing selection at these sites is notably lacking--indeed, whole-genome simulations indicate that our findings are expected under neutrality. We propose that (i) because it is rarely stable, long-term balancing selection is an evolutionary oddity, and (ii) when a balanced polymorphism is ancient in origin, the requirements for detection by means of SNP data alone will rarely be met.
Figures
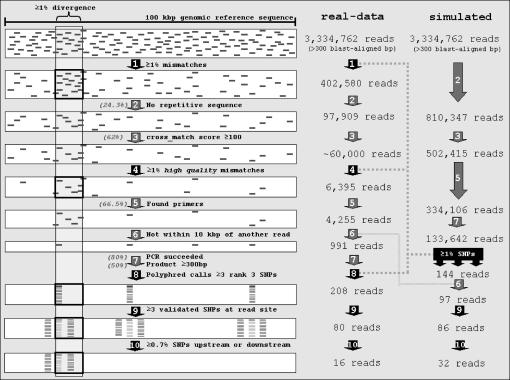
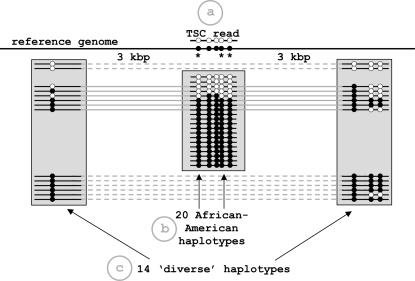

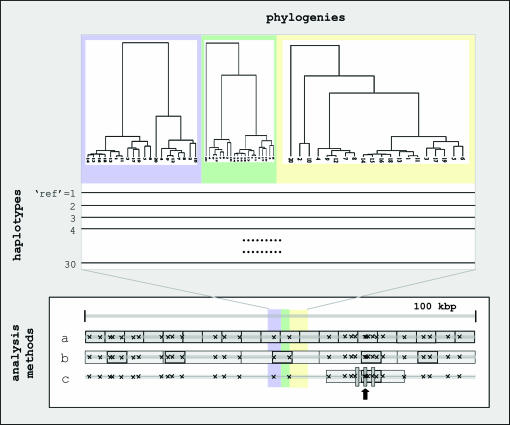
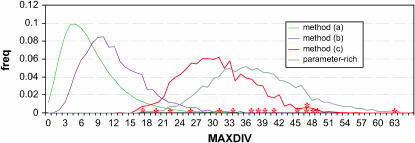


References
-
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. - PubMed
-
- Asthana, S., S. Schmidt and S. Sunyaev, 2005. A limited role for balancing selection. Trends Genet. 21: 30–32. - PubMed
-
- Barton, N. H., and A. Navarro, 2002. Extending the coalescent to multilocus systems: the case of balancing selection. Genet. Res. 79: 129–139. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
