

The origin and diversification of eukaryotes: problems with molecular phylogenetics and molecular clock estimation
- PMID: 16754613
- PMCID: PMC1578731
- DOI: 10.1098/rstb.2006.1845
The origin and diversification of eukaryotes: problems with molecular phylogenetics and molecular clock estimation
Abstract
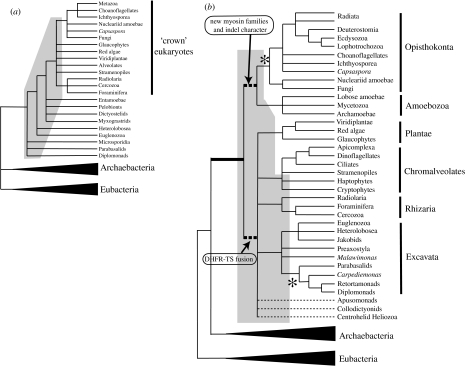
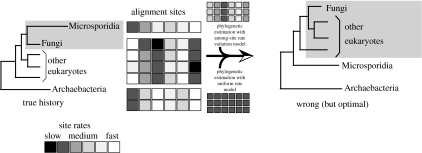
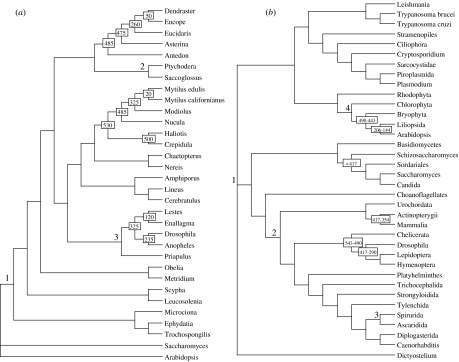
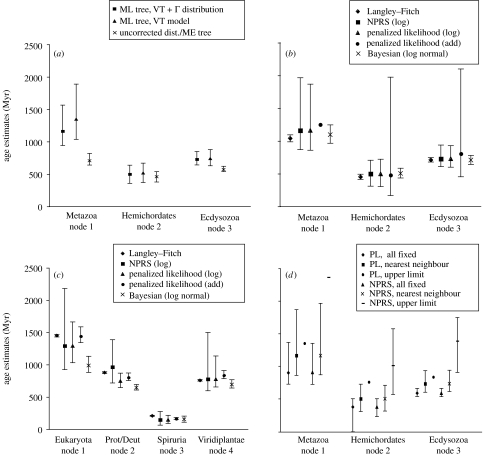
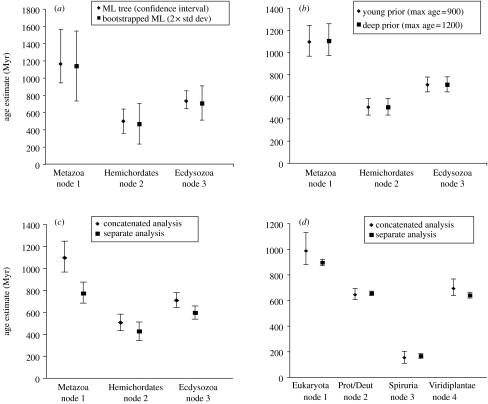
Determining the relationships among and divergence times for the major eukaryotic lineages remains one of the most important and controversial outstanding problems in evolutionary biology. The sequencing and phylogenetic analyses of ribosomal RNA (rRNA) genes led to the first nearly comprehensive phylogenies of eukaryotes in the late 1980s, and supported a view where cellular complexity was acquired during the divergence of extant unicellular eukaryote lineages. More recently, however, refinements in analytical methods coupled with the availability of many additional genes for phylogenetic analysis showed that much of the deep structure of early rRNA trees was artefactual. Recent phylogenetic analyses of a multiple genes and the discovery of important molecular and ultrastructural phylogenetic characters have resolved eukaryotic diversity into six major hypothetical groups. Yet relationships among these groups remain poorly understood because of saturation of sequence changes on the billion-year time-scale, possible rapid radiations of major lineages, phylogenetic artefacts and endosymbiotic or lateral gene transfer among eukaryotes. Estimating the divergence dates between the major eukaryote lineages using molecular analyses is even more difficult than phylogenetic estimation. Error in such analyses comes from a myriad of sources including: (i) calibration fossil dates, (ii) the assumed phylogenetic tree, (iii) the nucleotide or amino acid substitution model, (iv) substitution number (branch length) estimates, (v) the model of how rates of evolution change over the tree, (vi) error inherent in the time estimates for a given model and (vii) how multiple gene data are treated. By reanalysing datasets from recently published molecular clock studies, we show that when errors from these various sources are properly accounted for, the confidence intervals on inferred dates can be very large. Furthermore, estimated dates of divergence vary hugely depending on the methods used and their assumptions. Accurate dating of divergence times among the major eukaryote lineages will require a robust tree of eukaryotes, a much richer Proterozoic fossil record of microbial eukaryotes assignable to extant groups for calibration, more sophisticated relaxed molecular clock methods and many more genes sampled from the full diversity of microbial eukaryotes.
Figures
References
-
- Andersson J.O. Lateral gene transfer in eukaryotes. Cell. Mol. Life Sci. 2005;62:1182–1197. 10.1007/s00018-005-4539-z - DOI - PMC - PubMed
-
- Andersson J.O, Sarchfield S.W, Roger A.J. Gene transfers from Nanoarchaeota to an ancestor of diplomonads and parabasalids. Mol. Biol. Evol. 2005;22:85–90. 10.1093/molbev/msh254 - DOI - PubMed
-
- Aravind L, Koonin E.V. Eukaryotic-specific domains in translation initiation factors: implications for translation regulation and evolution of the translation system. Genome Res. 2000;10:1172–1184. 10.1101/gr.10.8.1172 - DOI - PMC - PubMed
-
- Archibald J.M. Jumping genes and shrinking genomes—probing the evolution of eukaryotic photosynthesis with genomics. IUBMB Life. 2005;57:539–547. - PubMed
-
- Archibald J.M, Longet D, Pawlowski J, Keeling P.J. A novel polyubiquitin structure in Cercozoa and Foraminifera: evidence for a new eukaryotic supergroup. Mol. Biol. Evol. 2003;20:62–66. 10.1093/molbev/msg006 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
