

Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity
- PMID: 16754847
- PMCID: PMC1482588
- DOI: 10.1073/pnas.0603503103
Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity
Abstract
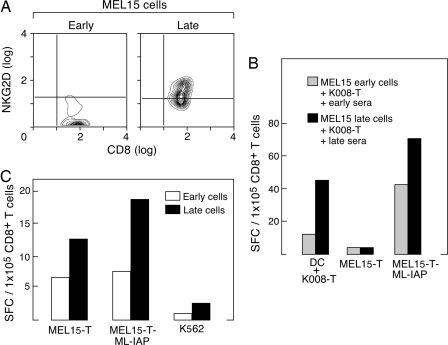
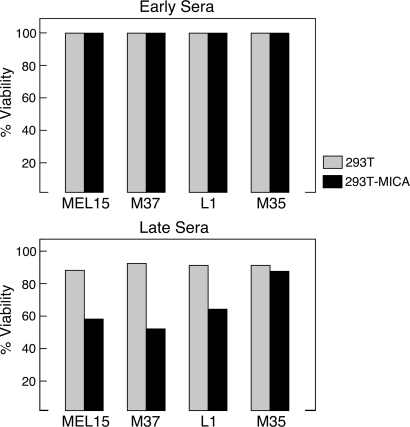
The activation of NKG2D on innate and adaptive cytotoxic lymphocytes contributes to immune-mediated tumor destruction. Nonetheless, tumor cell shedding of NKG2D ligands, such as MHC class I chain-related protein A (MICA), results in immune suppression through down-regulation of NKG2D surface expression. Here we show that some patients who respond to antibody-blockade of cytotoxic T lymphocyte-associated antigen 4 or vaccination with lethally irradiated, autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor generate high titer antibodies against MICA. These humoral reactions are associated with a reduction of circulating soluble MICA (sMICA) and an augmentation of natural killer (NK) cell and CD8(+) T lymphocyte cytotoxicity. The immunotherapy-induced anti-MICA antibodies efficiently opsonize cancer cells for dendritic cell cross-presentation, which is correlated with a diversification of tumor antigen recognition. The anti-MICA antibodies also accomplish tumor cell lysis through complement fixation. Together, these findings establish a key role for the NKG2D pathway in the clinical activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade and granulocyte-macrophage colony-stimulating factor secreting tumor cell vaccines. Moreover, these results highlight the therapeutic potential of anti-MICA antibodies to overcome immune suppression and effectuate tumor destruction in patients.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
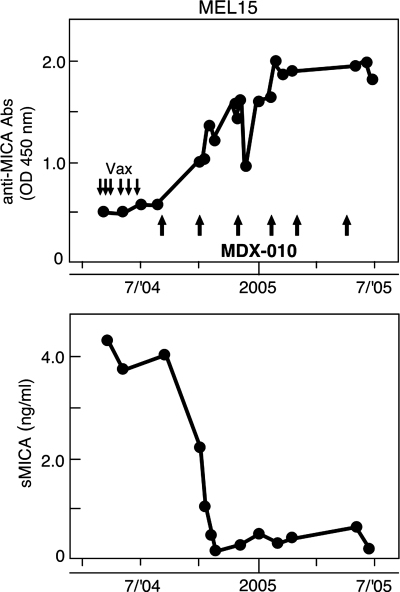
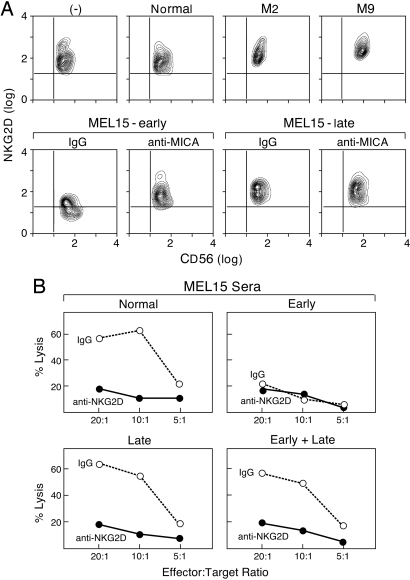
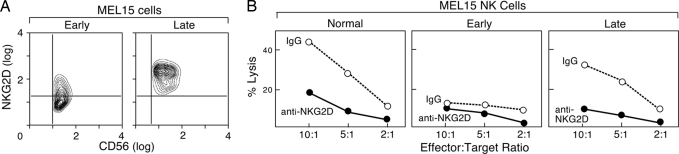
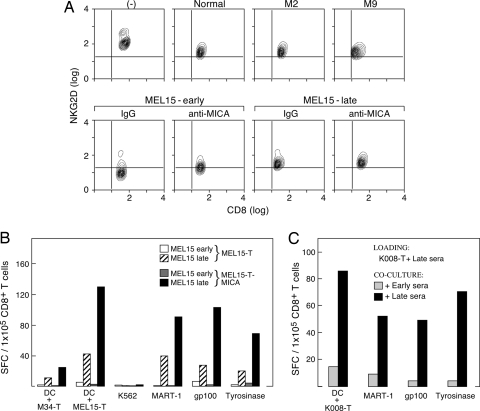
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
