

Rac1 links integrin-mediated adhesion to the control of lactational differentiation in mammary epithelia
- PMID: 16754961
- PMCID: PMC2063893
- DOI: 10.1083/jcb.200601059
Rac1 links integrin-mediated adhesion to the control of lactational differentiation in mammary epithelia
Abstract
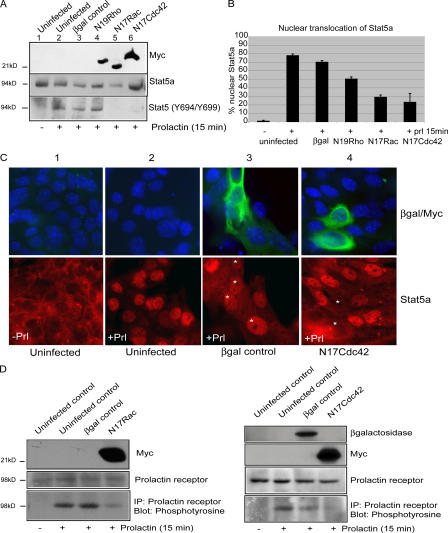
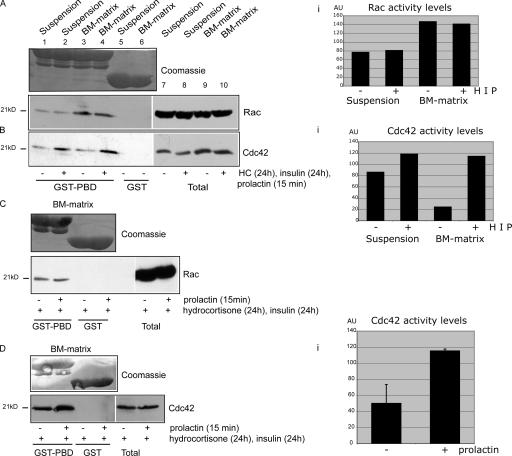
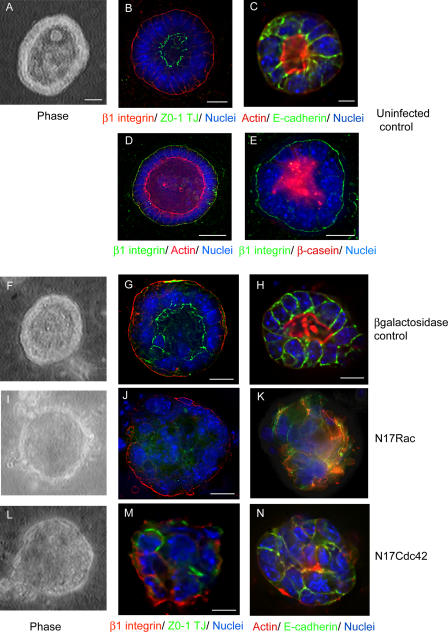
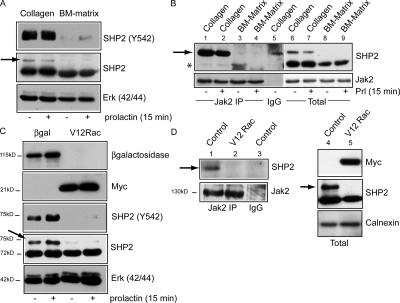
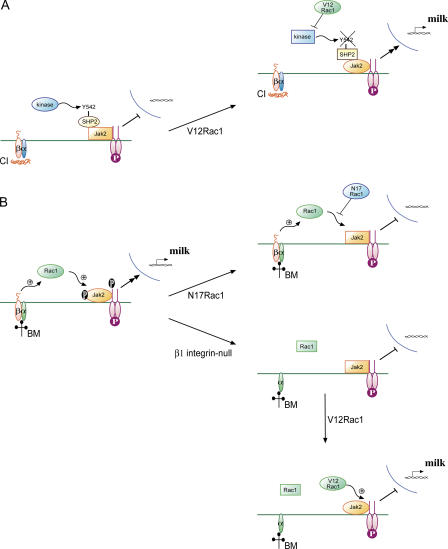
The expression of tissue-specific genes during mammary gland differentiation relies on the coincidence of two distinct signaling events: the continued engagement of beta1 integrins with the extracellular matrix (ECM) and a hormonal stimulus from prolactin (Prl). How the integrin and Prl receptor (PrlR) systems integrate to regulate milk protein gene synthesis is unknown. In this study, we identify Rac1 as a key link. Dominant-negative Rac1 prevents Prl-induced synthesis of the milk protein beta-casein in primary mammary epithelial cells cultured as three-dimensional acini on basement membrane. Conversely, activated Rac1 rescues the defective beta-casein synthesis that occurs under conditions not normally permissive for mammary differentiation, either in beta1 integrin-null cells or in wild-type cells cultured on collagen. Rac1 is required downstream of integrins for activation of the PrlR/Stat5 signaling cascade. Cdc42 is also necessary for milk protein synthesis but functions via a distinct mechanism to Rac1. This study identifies the integration of signals provided by ECM and hormones as a novel role for Rho family guanosine triphosphatases.
Figures




References
-
- Aggeler, J., J. Ward, L.M. Blackie, M.H. Barcellos-Hoff, C.H. Streuli, and M.J. Bissell. 1991. Cytodifferentiation of mouse mammary epithelial cells cultured on a reconstituted basement membrane reveals striking similarities to development in vivo. J. Cell Sci. 99:407–417. - PubMed
-
- Benitah, S.A., M. Frye, M. Glogauer, and F.M. Watt. 2005. Stem cell depletion through epidermal deletion of Rac1. Science. 309:933–935. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
