

The molecular biology of thrombotic microangiopathy
- PMID: 16760911
- PMCID: PMC2497001
- DOI: 10.1038/sj.ki.5001535
The molecular biology of thrombotic microangiopathy
Abstract
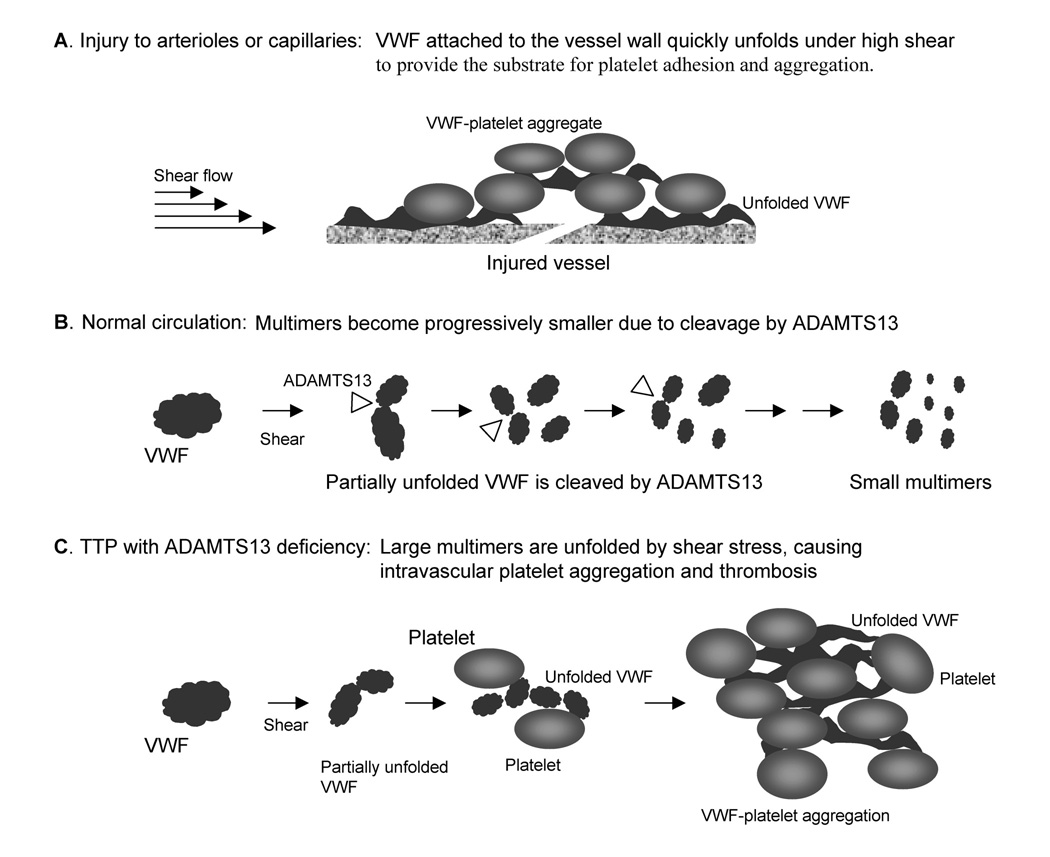
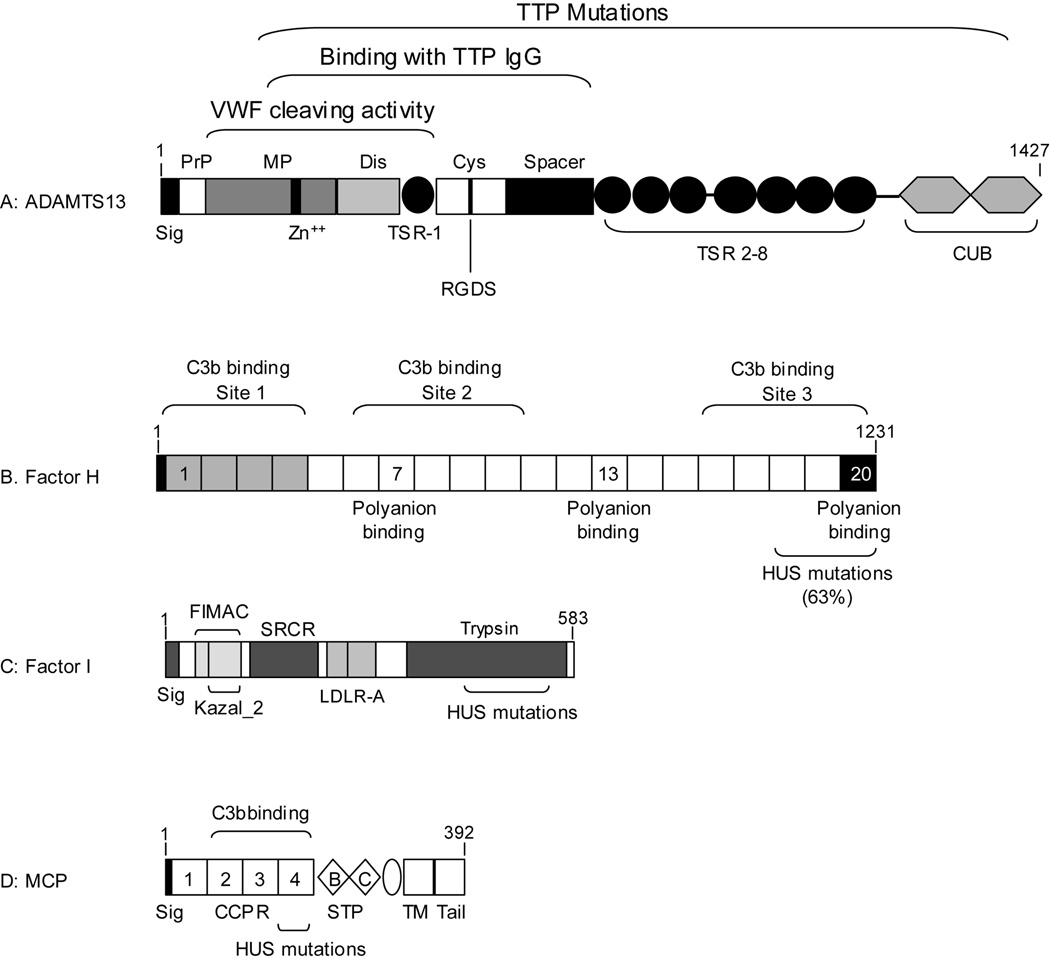
Thrombotic microangiopathy, which includes thrombotic thrombocytopenic purpura (TTP), shiga-toxin-associated hemolytic uremic syndrome (Stx-HUS) and atypical HUS, is characterized by the development of hyaline thrombi in the microvasculature resulting in thrombocytopenia, microangiopathic hemolysis, and organ dysfunction. Renal failure is a predominant complication of both Stx-HUS and atypical HUS, whereas neurological complications are more prominent in TTP. Other disorders such as lupus or bone marrow transplantations may occasionally present with features of thrombotic microangiopathy. Recent studies have found autoimmune inhibitors or genetic mutations of a von Willebrand factor (VWF) cleaving metalloprotease ADAMTS13 in patients with TTP. In approximately 30-50% of patients with atypical HUS, mutations have been detected in complement factor H, membrane cofactor protein (CD46), or factor I. All three proteins are involved in the regulation of complement activation. Additionally, autoantibodies of factor H have been described in patients without genetic mutations. These advances illustrate that dysregulation of VWF homeostasis or complement activation owing to genetic or autoimmune mechanisms may lead to the syndrome of thrombotic microangiopathy.
Figures

References
-
- Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
