

DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy
- PMID: 16773573
- PMCID: PMC1474134
- DOI: 10.1086/504393
DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy
Abstract
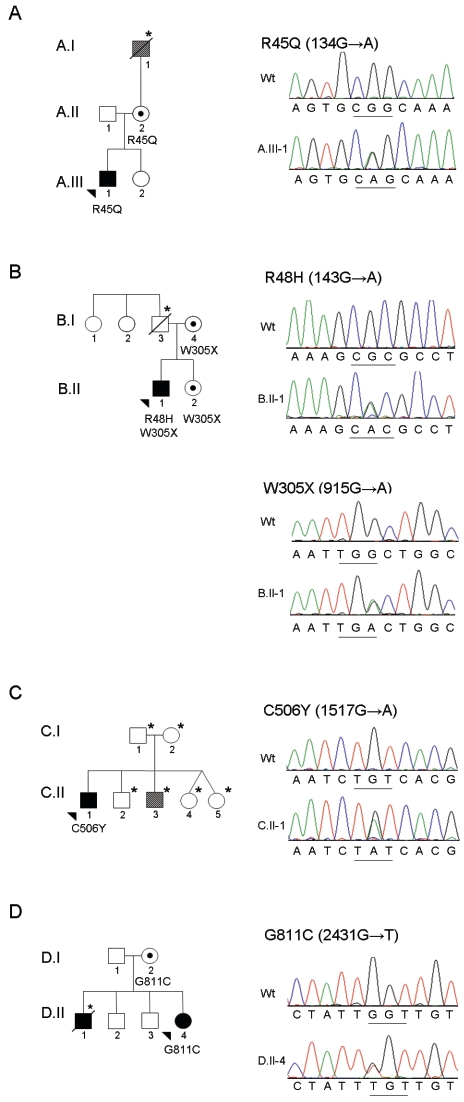
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is a disorder characterized by fibrofatty replacement of cardiac myocytes that typically manifests in the right ventricle. It is inherited as an autosomal dominant disease with reduced penetrance, although autosomal recessive forms of the disease also occur. We identified four probands with ARVD/C caused by mutations in DSG2, which encodes desmoglein-2, a component of the cardiac desmosome. No association between mutations in this gene and human disease has been reported elsewhere. One of these probands has compound-heterozygous mutations in DSG2, and the remaining three have isolated heterozygous missense mutations, each disrupting known functional components of desmoglein-2. We report that mutations in DSG2 contribute to the development of ARVD/C.
Figures


References
Web Resources
-
- Johns Hopkins ARVD Program, http://www.arvd.com/
-
- Online Mendelian Inheritance in Man (OMIM): http://www.ncbi.nlm.nih.gov/Omim/ (for ARVD/C, Naxos syndrome, and Carvajal syndrome)
References
-
- McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F, for the Task Force of the Working Group on Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology (1994) Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J 71:215–218 - PMC - PubMed
-
- Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA (2002) Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 71:1200–1206 - PMC - PubMed
-
- McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355:2119–2124 10.1016/S0140-6736(00)02379-5 - DOI - PubMed
-
- Norgett EE, Hatsell SJ, Carvajal-Huerta L, Ruiz Cabezas J-C, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP (2000) Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 9:2761–2766 10.1093/hmg/9.18.2761 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
