

p53 downregulates its activating vaccinia-related kinase 1, forming a new autoregulatory loop
- PMID: 16782868
- PMCID: PMC1489172
- DOI: 10.1128/MCB.00069-06
p53 downregulates its activating vaccinia-related kinase 1, forming a new autoregulatory loop
Abstract
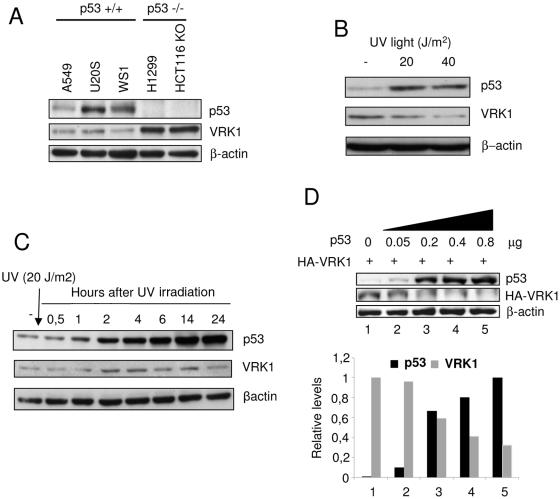
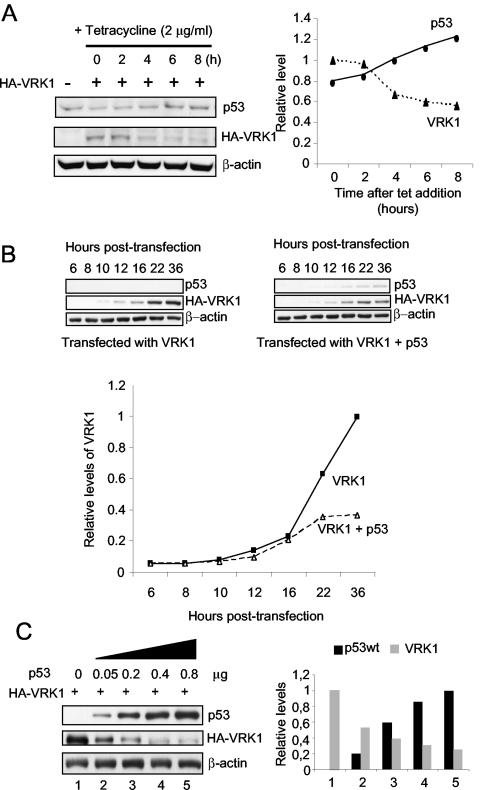
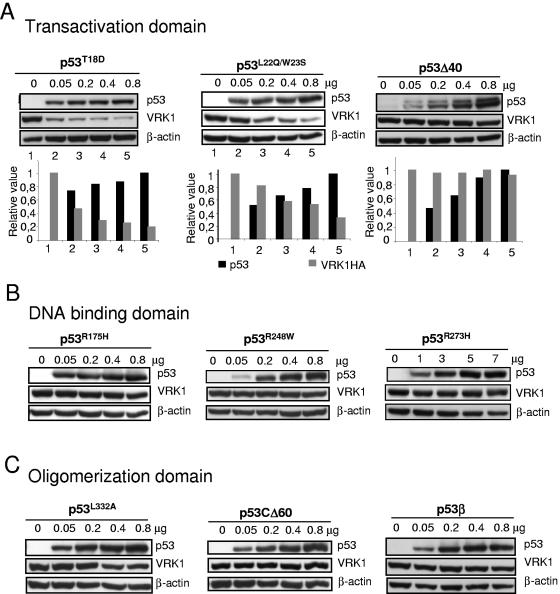
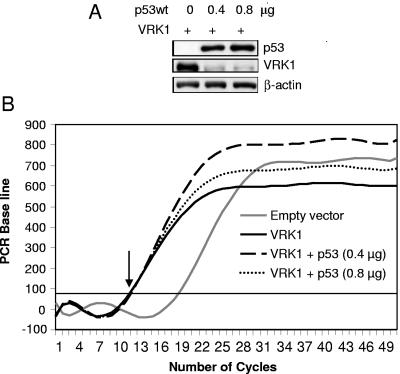
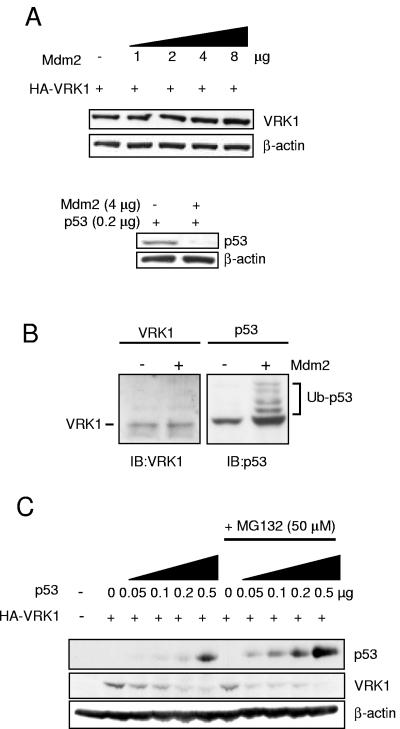
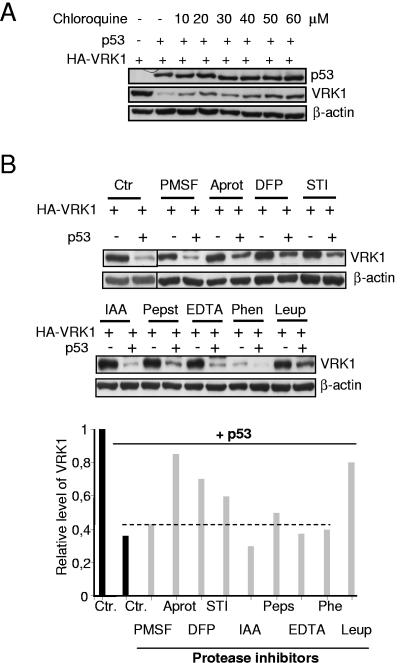
The stable accumulation of p53 is detrimental to the cell because it blocks cell growth and division. Therefore, increases in p53 levels are tightly regulated, mainly by its transcriptional target, mdm2, that downregulates p53. Elucidation of new signaling pathways requires the characterization of the members and the nature of their connection. Vaccinia-related kinase 1 (VRK1) contributes to p53 stabilization by partly interfering with its mdm2-mediated degradation, among other mechanisms; therefore, it is likely that some form of autoregulation between VRK1 and p53 must occur. We report here the identification of an autoregulatory loop between p53 and its stabilizing VRK1. There is an inverse correlation between VRK1 and p53 levels in cell lines, and induction of p53 by UV light downregulates VRK1 in fibroblasts. As the amount of p53 protein increases, there is a downregulation of the VRK1 protein level independent of its promoter. This effect is indirect but requires a transcriptionally active p53. The three most common transcriptionally inactive mutations detected in hereditary (Li-Fraumeni syndrome) and sporadic human cancer, p53(R175H), p53(R248W), and p53(R273H), as well as p53(R280K), are unable to induce downregulation of VRK1 protein. The p53 isoforms Delta40p53 and p53beta, lacking the transactivation and oligomerization domains, respectively, do not downregulate VRK1. VRK1 downregulation induced by p53 is independent of mdm2 activity and proteasome-mediated degradation since it occurs in the presence of proteasome inhibitors and in mdm2-deficient cells. The degradation of VRK1 is sensitive to chloroquine, an inhibitor of the late endosome-lysosome transport, and to serine protease inhibitors of the lysosomal pathway.
Figures

References
-
- Agarwal, M. L., W. R. Taylor, M. V. Chernov, O. B. Chernova, and G. R. Stark. 1998. The p53 network. J. Biol. Chem. 273:1-4. - PubMed
-
- Barcia, R., S. Lopez-Borges, F. M. Vega, and P. A. Lazo. 2002. Kinetic properties of p53 phosphorylation by the human vaccinia-related kinase 1. Arch. Biochem. Biophys. 399:1-5. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous