

Inheritance pattern of familial moyamoya disease: autosomal dominant mode and genomic imprinting
- PMID: 16788009
- PMCID: PMC2077755
- DOI: 10.1136/jnnp.2006.096040
Inheritance pattern of familial moyamoya disease: autosomal dominant mode and genomic imprinting
Abstract
Background: Although the aetiology of moyamoya disease (MMD) has not been fully clarified, genetic analysis of familial MMD (F-MMD) has considerable potential to disclose it.
Objective: To determine the inheritance pattern and clinical characteristics of F-MMD to enable precise genetic analyses of the disease.
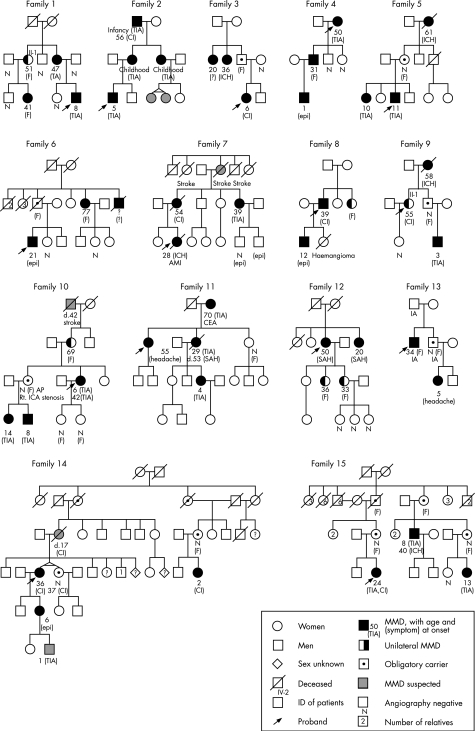
Methods: 15 highly aggregated Japanese families (52 patients; 38 women and 14 men) with three or more affected members were examined. The difference in categories of age at onset (child onset, adult onset and asymptomatic) between paternal and maternal transmission was compared by chi2 statistics.
Results: In all families there had been three or more generations without consanguinity, and all types of transmission, including father-to-son, were observed. Among a total of 135 offspring of affected people, 59 (43.7%) were patients with MMD or obligatory carriers. Affected mothers were more likely to produce late-onset (adult-onset or asymptomatic) female offspring (p = 0.007).
Conclusions: The mode of inheritance of F-MMD is autosomal dominant with incomplete penetrance. Thus, in future genetic studies on F-MMD, parametric linkage analyses using large families with an autosomal dominant mode of inheritance are recommended. Genomic imprinting may be associated with the disease.
Conflict of interest statement
Competing interests: None declared.
References
-
- Wakai K, Tamakoshi A, Ikezaki K.et al Epidemiological features of moyamoya disease in Japan: findings from a nationwide survey. Clin Neurol Neurosurg 199799(Suppl 2)P1–P5. - PubMed
-
- Yonekawa Y, Ogata N, Kaku Y.et al Moyamoya disease in Europe, past and present status. Clin Neurol Neurosurg 199799(Suppl 2)P58–P60. - PubMed
-
- Tsuji I, Kuriyama S, Kusaka Y.et al Epidemiological and clinical analyses of moyamoya disease in 2004. Annual report 2004 of the Research Committee on Moyamoya Disease (Spontaneous Occlusion of the Circle of Willis) of Health and Labour Sciences Research Grants, Research on measures for intractable diseases 200513–17.
-
- Fukui M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (‘moyamoya' disease): Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare, Japan. Clin Neurol Neurosurg 199799(Suppl 2)P238–P240. - PubMed
-
- Houkin K. Novel MRA staging of moyamoya disease. Annual report 2001 of the Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of Ministry of Health Labour and Welfare 2002:39–43 (In Japanese with English abstract)
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources