

Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations
- PMID: 16789823
- PMCID: PMC1513265
- DOI: 10.1371/journal.pgen.0020116
Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations
Abstract
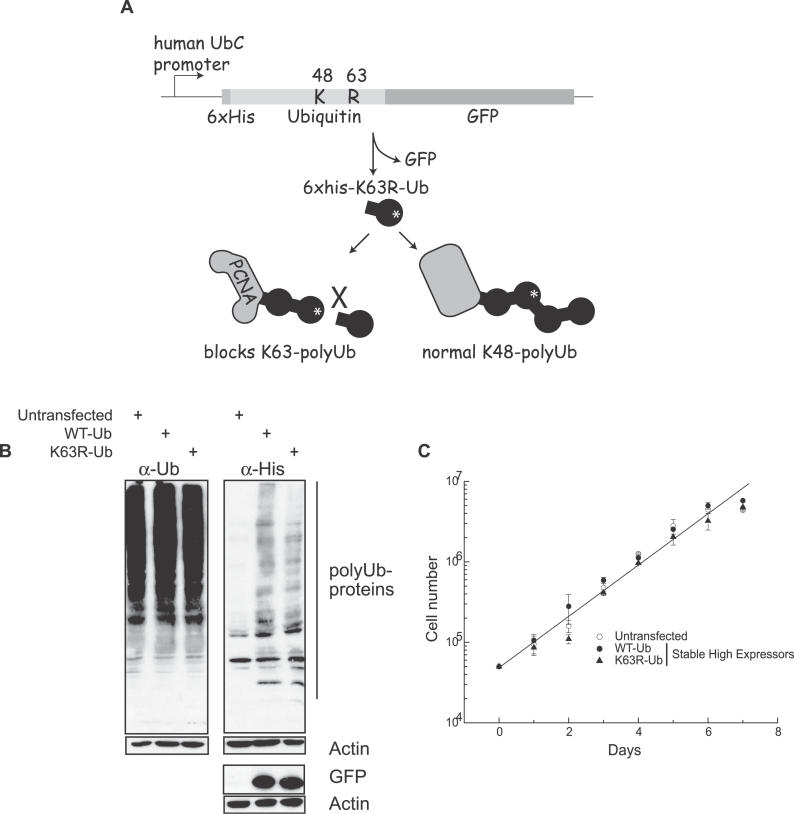
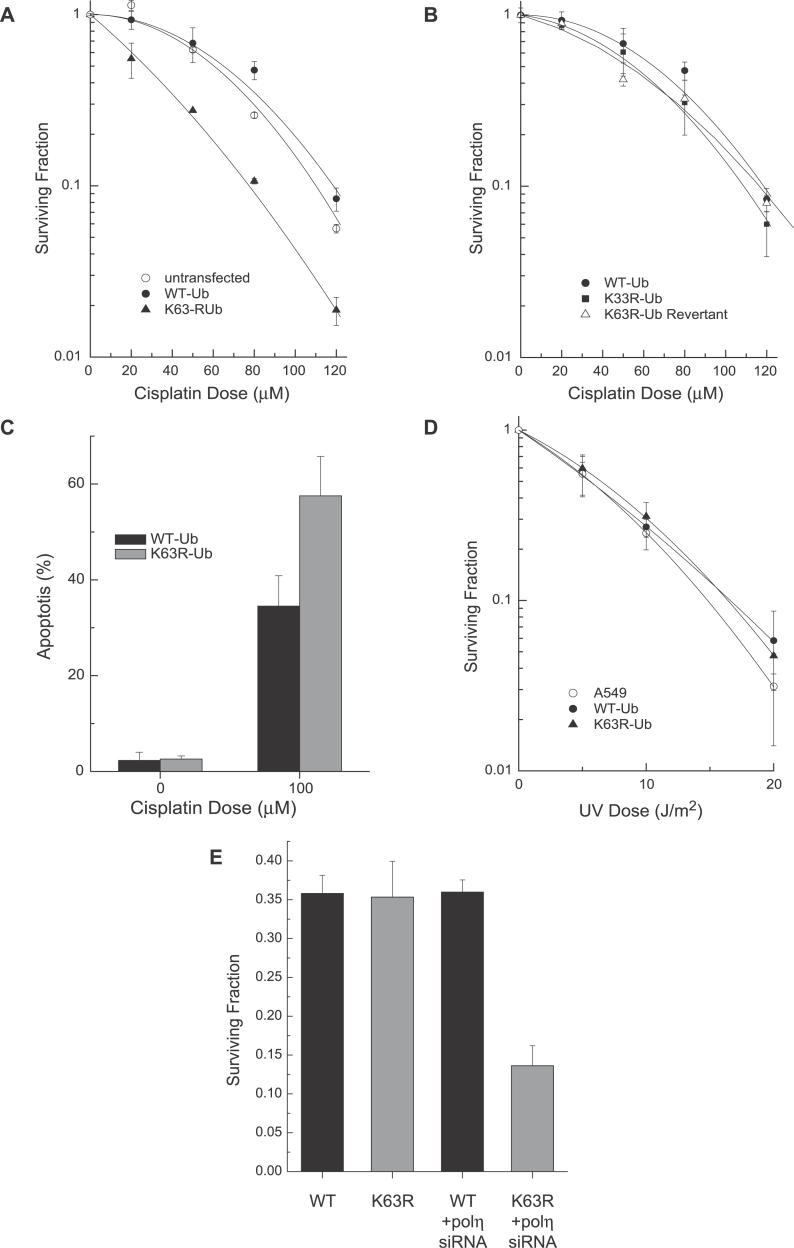
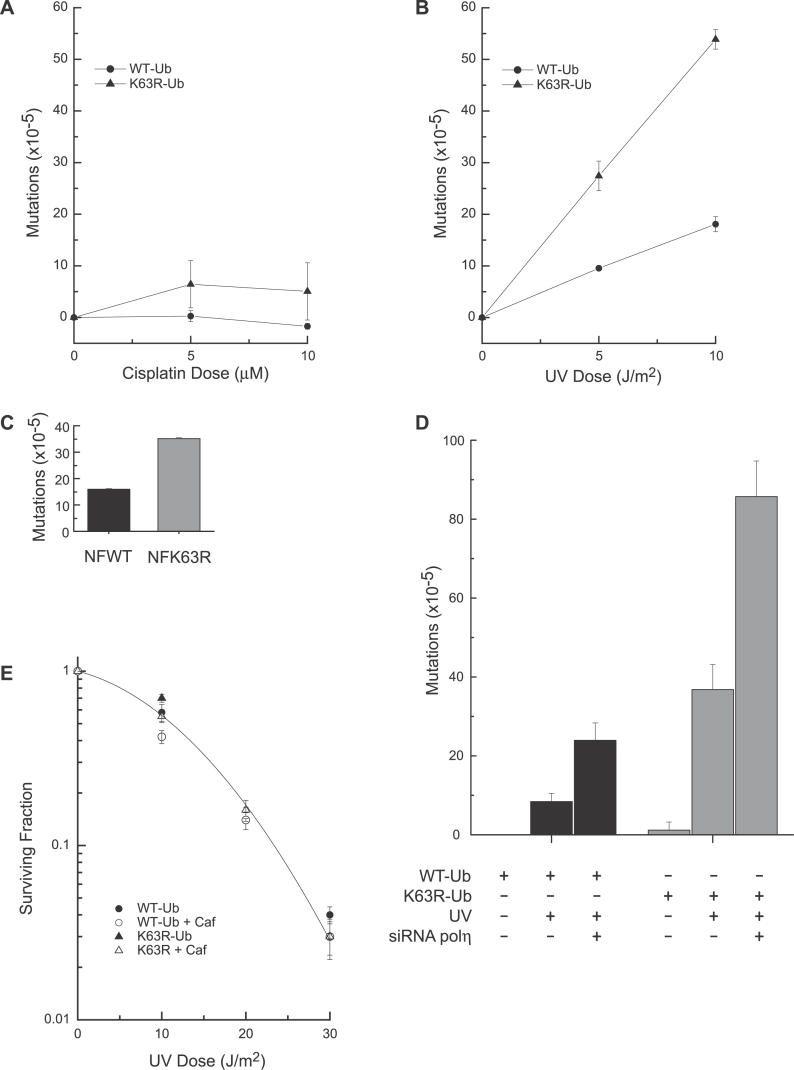
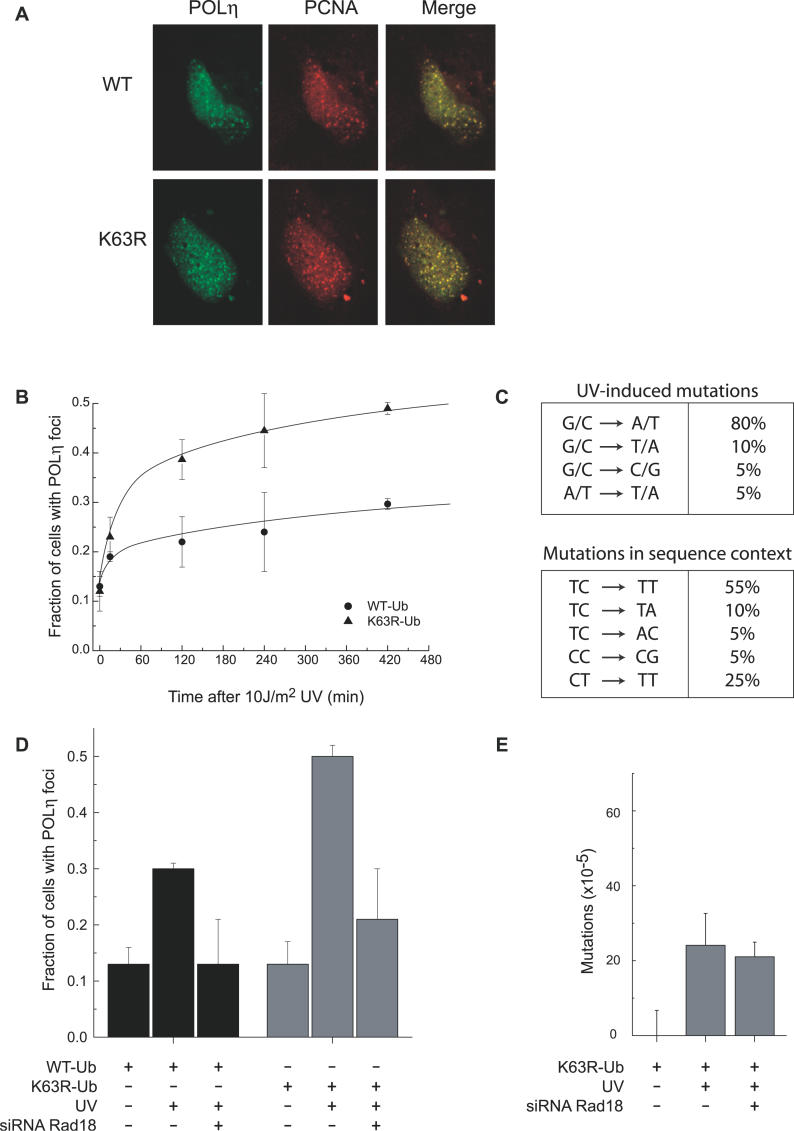
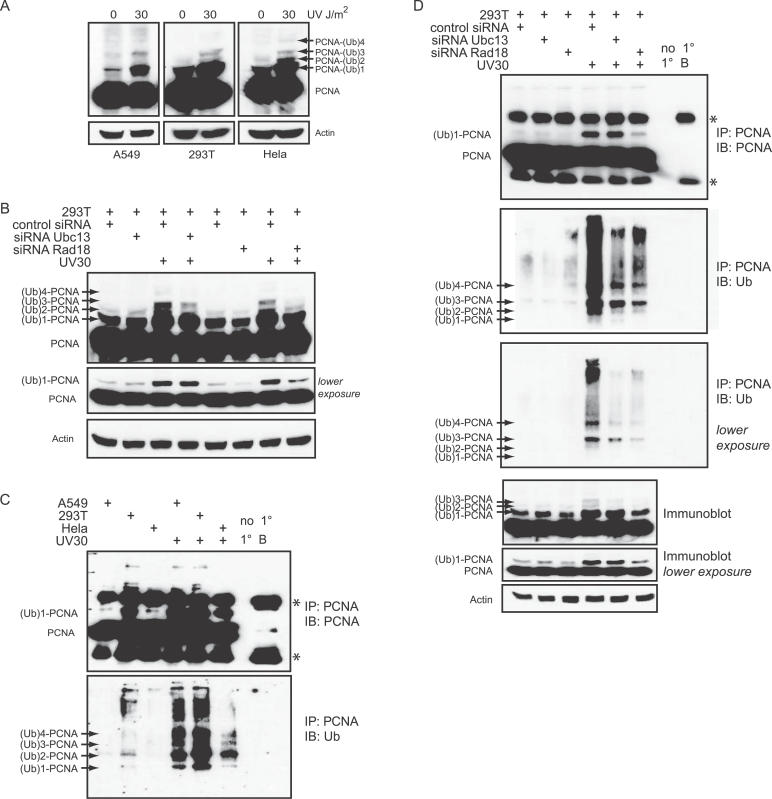
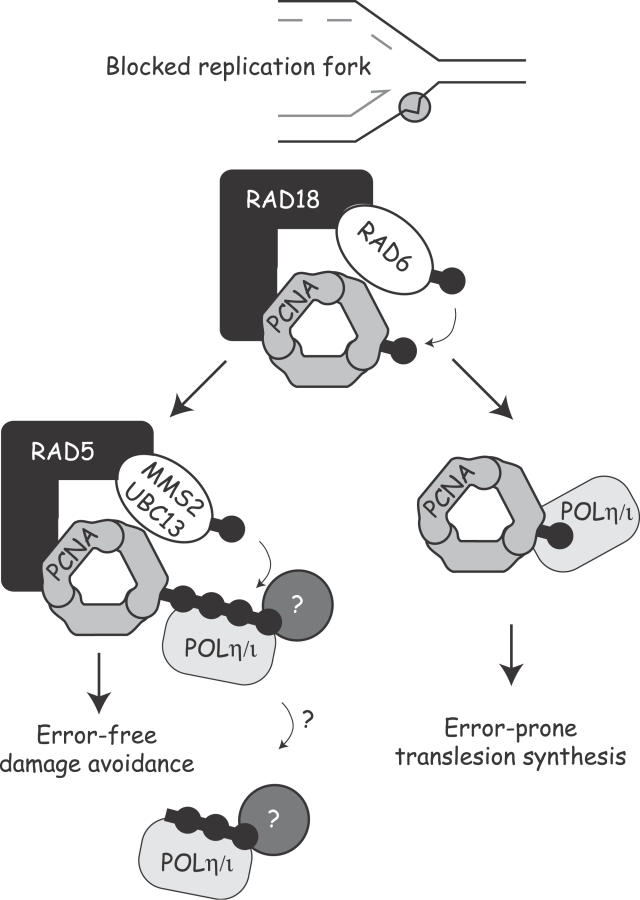
Eukaryotic cells possess several mechanisms to protect the integrity of their DNA against damage. These include cell-cycle checkpoints, DNA-repair pathways, and also a distinct DNA damage-tolerance system that allows recovery of replication forks blocked at sites of DNA damage. In both humans and yeast, lesion bypass and restart of DNA synthesis can occur through an error-prone pathway activated following mono-ubiquitination of proliferating cell nuclear antigen (PCNA), a protein found at sites of replication, and recruitment of specialized translesion synthesis polymerases. In yeast, there is evidence for a second, error-free, pathway that requires modification of PCNA with non-proteolytic lysine 63-linked polyubiquitin (K63-polyUb) chains. Here we demonstrate that formation of K63-polyUb chains protects human cells against translesion synthesis-induced mutations by promoting recovery of blocked replication forks through an alternative error-free mechanism. Furthermore, we show that polyubiquitination of PCNA occurs in UV-irradiated human cells. Our findings indicate that K63-polyubiquitination guards against environmental carcinogenesis and contributes to genomic stability.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures
References
-
- Spivak G, Hanawalt PC. Translesion DNA synthesis in the dihydrofolate reductase domain of UV-irradiated CHO cells. Biochemistry. 1992;31:6794–6800. - PubMed
-
- Wu HI, Brown JA, Dorie MJ, Lazzeroni L, Brown JM. Genome-wide identification of genes conferring resistance to the anticancer agents cisplatin, oxaliplatin, and mitomycin C. Cancer Res. 2004;64:3940–3948. - PubMed
-
- Simon JA, Szankasi P, Nguyen DK, Ludlow C, Dunstan HM, et al. Differential toxicities of anticancer agents among DNA repair and checkpoint mutants of Saccharomyces cerevisiae . Cancer Res. 2000;60:328–333. - PubMed
-
- Bailly V, Lauder S, Prakash S, Prakash L. Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J Biol Chem. 1997;272:23360–23365. - PubMed
-
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
