

Review
doi: 10.1016/j.drudis.2006.05.012.
Virtual ligand screening: strategies, perspectives and limitations
Affiliations
- PMID: 16793526
- PMCID: PMC7108249
- DOI: 10.1016/j.drudis.2006.05.012
Item in Clipboard
Review
Virtual ligand screening: strategies, perspectives and limitations
Drug Discov Today.
2006 Jul.
Abstract
In contrast to high-throughput screening, in virtual ligand screening (VS), compounds are selected using computer programs to predict their binding to a target receptor. A key prerequisite is knowledge about the spatial and energetic criteria responsible for protein-ligand binding. The concepts and prerequisites to perform VS are summarized here, and explanations are sought for the enduring limitations of the technology. Target selection, analysis and preparation are discussed, as well as considerations about the compilation of candidate ligand libraries. The tools and strategies of a VS campaign, and the accuracy of scoring and ranking of the results, are also considered.
Figures
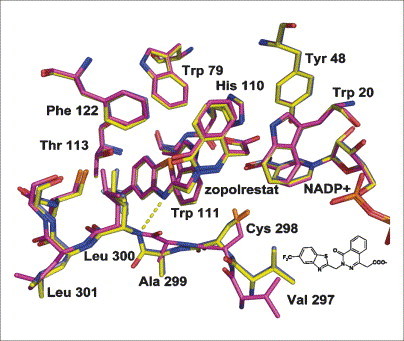
Binding mode differs with soaking and co-crystallization conditions. Crystal structure of aldose reductase with the inhibitor zopolrestat is shown. The crystal structure determined after one day of soaking (yellow) differs from the structure obtained after six days of soaking (magenta) but is identical to that obtained by cocrystallization (light blue). Whereas in the one day-soaked structure (yellow) the amide bond between Ala299 and Leu300 orientates its NH group towards the inhibitor to form a hydrogen bond (dotted yellow line) with the bound ligand, in the latter two structures (magenta and light blue) this amide bond rotates away from the inhibitor and no such hydrogen bond is observed .
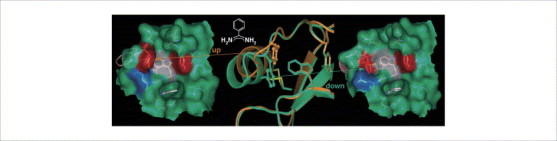
Induced-fit adaptations in two different crystal forms. Two crystal structures of benzamidine (in both cases, accommodated deeply buried in the S1 pocket, which is indicated as a deep depression in the left- and right-hand images) bound to a trypsin mutant exhibiting a binding pocket related to factor Xa. Both crystal structures differ in terms of the conformational state of Phe174 (in the left- and right-hand images at the left rim of the binding pocket with the red surface), which is exposed (‘up’) in one structure (orange and on the left) and buried (‘down’) in the other (green and on the right). This adaptation involves partial unwinding of a short three-turn helix (in the centre, helix at the left-hand side of the binding pocket) and rearrangement of a disulfide bond (in the centre, yellow bond to the back) , .
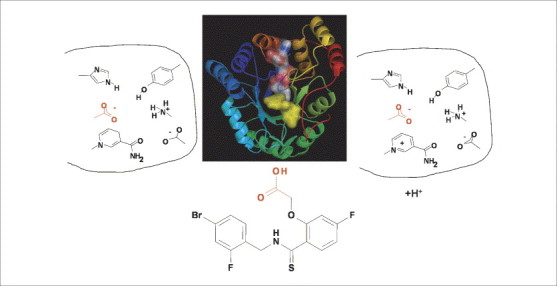
Protonation states can change upon ligand binding. Depending on the oxidation state of the bound cofactor NADPH (left) and NADP+ (right), aldose reductase binds to a carboxylate-type inhibitor (e.g. IDD594) by placing its acid functional group (red) into the catalytic centre; the complex thereby formed remains either in an unchanged protonation state (left) or picks up protons upon binding (right). In the centre, the crystal structure of the complex is shown. A short contact distance between the carboxylate function of the inhibitor (shown with a yellow surface) and the nicotinamide portion of the cofactor (shown with an atom-type coloured surface, where oxygen is red, nitrogen is blue, carbon is white, sulfur is yellow and phosphorous is orange) is formed. High resolution X-ray crystal structure analysis and neutron diffraction has provided evidence that the inhibitor binds with its carboxylate function in the deprotonated state, and the active site His is present in the neutral state. It remains unresolved where the proton goes in the case of the NADP+-bound complex, or whether the residues of the catalytic centre exhibit deviating protonation states in the uncomplexed and inhibited situation. With respect to VS, a unique and precise assignment of the protonation states of the ligand and protein functional groups in a pKa range between 3 and 11 is essential because, for example, for docking it is important whether such a group is considered as donor or acceptor of a hydrogen bond (Krämer and Klebe, unpublished).
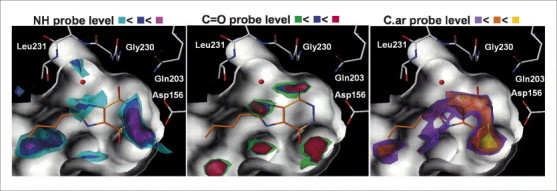
Mapping the hot spots of binding. Hotspot analysis using DrugScore for the binding pocket of t-RNA guanine transglycosylase (surface of the binding pocket indicated in white). Regions energetically favourable for the binding of a hydrogen bond donor group (represented by an NH group) or an acceptor group (represented by a C=O group), or a hydrophobic molecular portion (represented by aromatic carbon atoms, C.ar) have been analysed and contoured on three subsequent levels (indicated using three different colours) above the deepest energy minimum found for each atom type in the maps . The crystallographically determined binding mode of an inhibitor is shown superimposed.
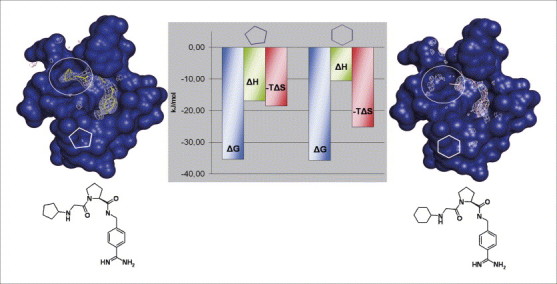
Similar ligands decompose differently into enthalpic and entropic binding contributions. Crystal structures of two closely related thrombin inhibitors bearing a cyclopentyl or cyclohexyl moiety as terminal substituent to accommodate the S3/S4 pocket of the catalytic site (surface of the binding pocket is indicated in blue). Whereas the five-membered ring (left) gives rise to a well-defined difference in electron density (white ‘chicken-wire’ contouring), the six-membered ring (right) cannot be assigned to any density (see inside white circles). It is likely that the latter fragment shows enhanced residual mobility and is scattered around several conformational states. Interestingly, this deviating behaviour is well reflected in the thermodynamic properties (centre). Both compounds exhibit the same free energy of binding (ΔG, blue columns). However, the cyclohexyl derivative (right) with the enhanced residual mobility is entropically (-TΔS, red columns) more favoured than the ‘less-well clamped’ five-ring derivative (left). The latter experiences better enthalpic contributions (ΔH, green columns) to binding .
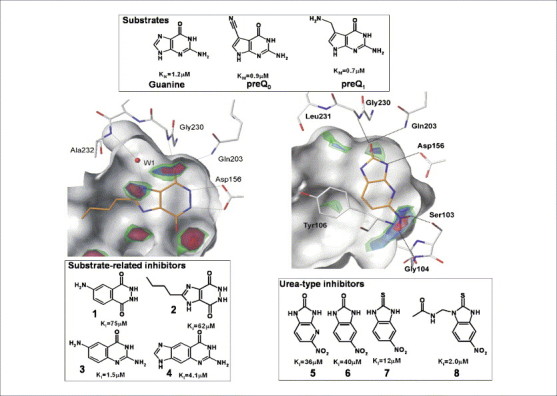
An unexpected ligand skeleton from virtual screening. Virtual screening (VS) has been used to search for putative inhibitors of t-RNA guanine transglycosylase . Initial hits such as 1–4 (lower left box), which were followed up by chemical synthesis, showed structural similarity with the natural substrates of this enzyme guanine, preQ0 and preQ1 (upper box). These searches were based on the protein-based pharmacophore shown on the left (contours for hydrogen bond acceptor group) and in Figure 4. An experienced medicinal chemist could possibly have come up with similar suggestions for potential leads such as 1–4. However, in a second VS campaign we focused on the replacement of two water molecules at the lower right rim of the binding pocket of t-RNA guanine transglycosylase (right, contours for hydrogen bond acceptor group [126]). This screen suggested 5 as a potential hit. Its cyclic urea-type skeleton is distinct in its structure and binding mode from any known natural substrate and it can serve as a synthetically easily accessible lead. Several derivatives (e.g. 6–8) have been synthesized and show a reasonable structure–activity relationship (Stengl et al., unpublished). It is unlikely that this novel lead structure would have been suggested by comparative substrate considerations. This example underlines the power of VS as an alternative source for novel lead discovery.
References
-
- Bolten B.M., DeGregorio T. Trends in development cycles. Nat. Rev. Drug Discov. 2002;1:335–336. - PubMed
-
- Fishman M.C., Porter J.A. A new grammar for drug discovery. Nature. 2005;437:491–493. - PubMed
-
- Lahana R. How many leads from HTS? Drug Discov. Today. 1999;4:447–448. - PubMed
-
- Ramesha C.S. Comment: How many leads from HTS? Drug Discov. Today. 2000;5:43–44. - PubMed
-
- Langer T., Krovat E.M. Chemical feature-based pharmacophores and virtual library screening for discovery of new leads. Curr. Opin. Drug Discov. Devel. 2003;6:370–376. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
