

Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain
- PMID: 16793892
- PMCID: PMC4758341
- DOI: 10.1523/JNEUROSCI.5444-05.2006
Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain
Abstract
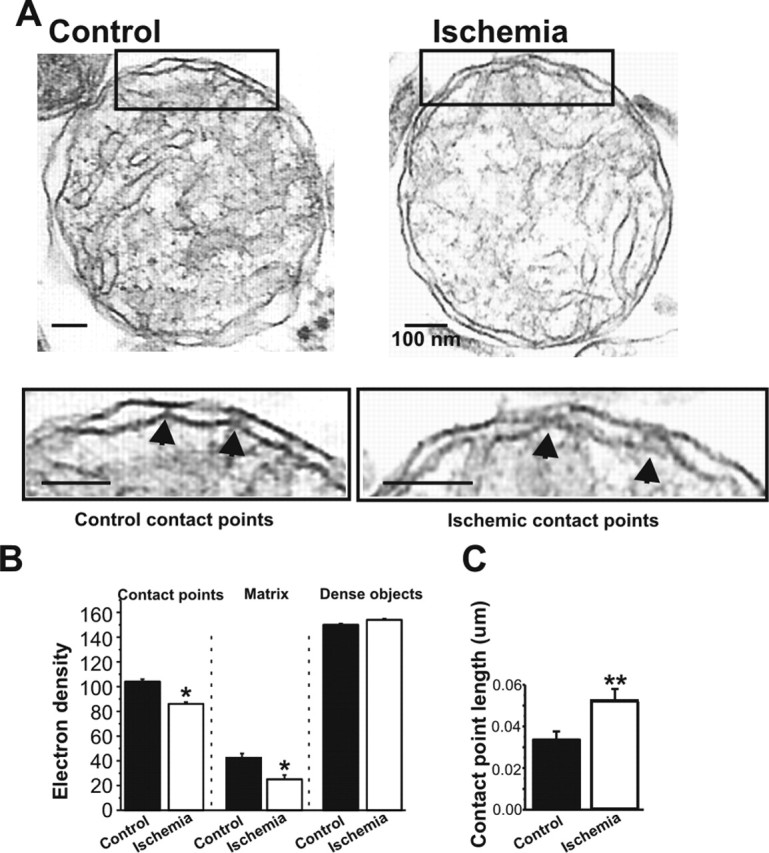
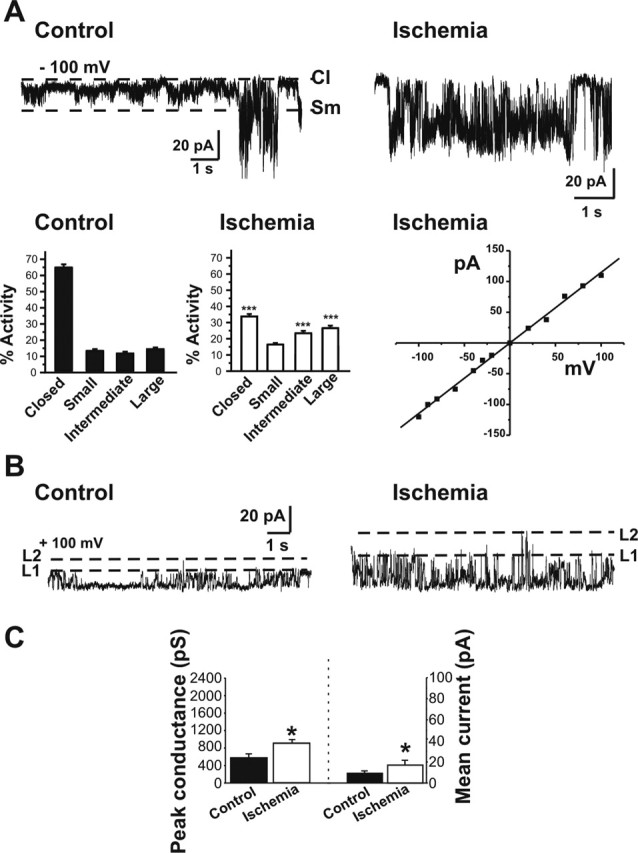
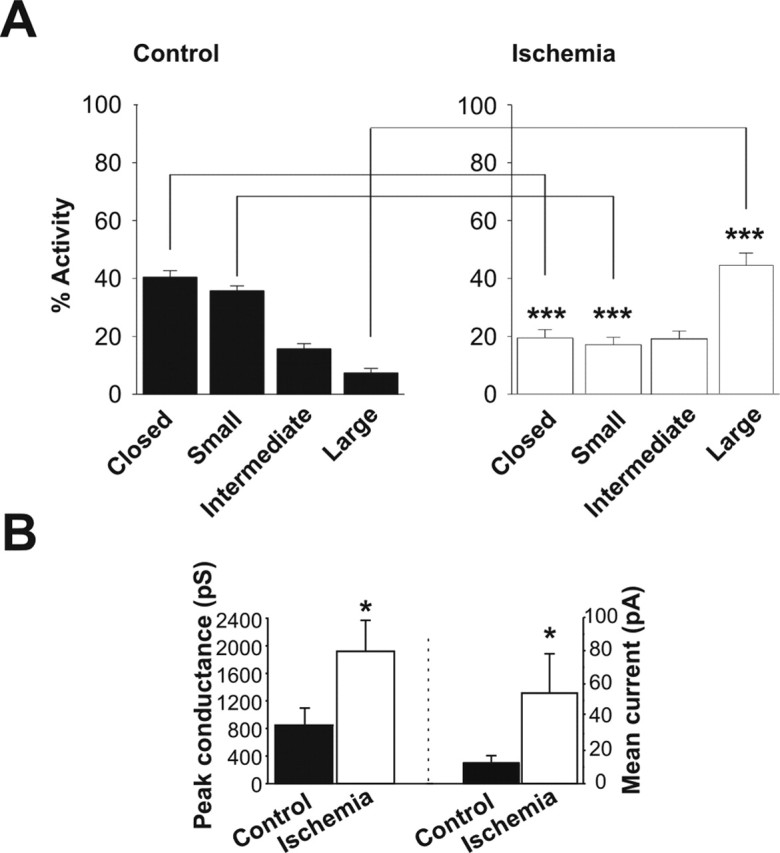
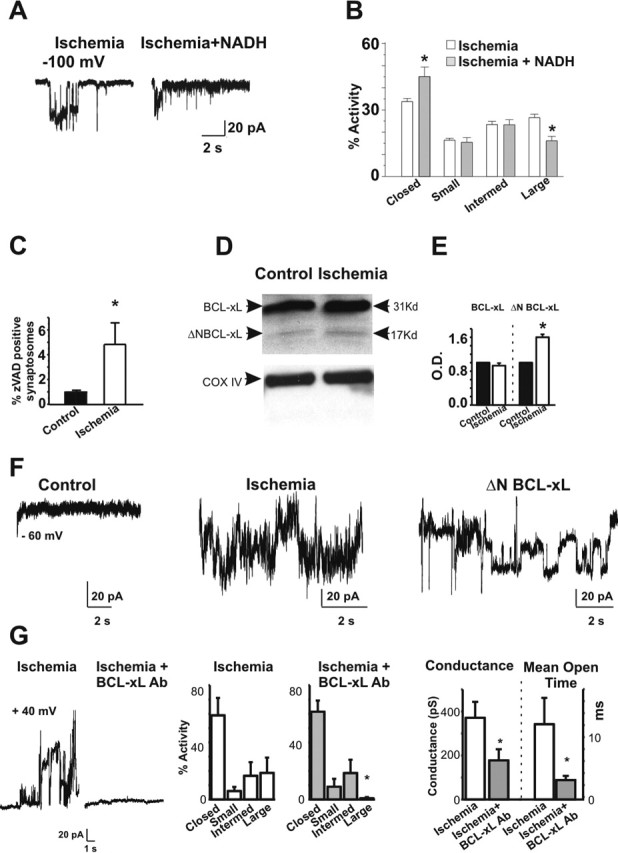
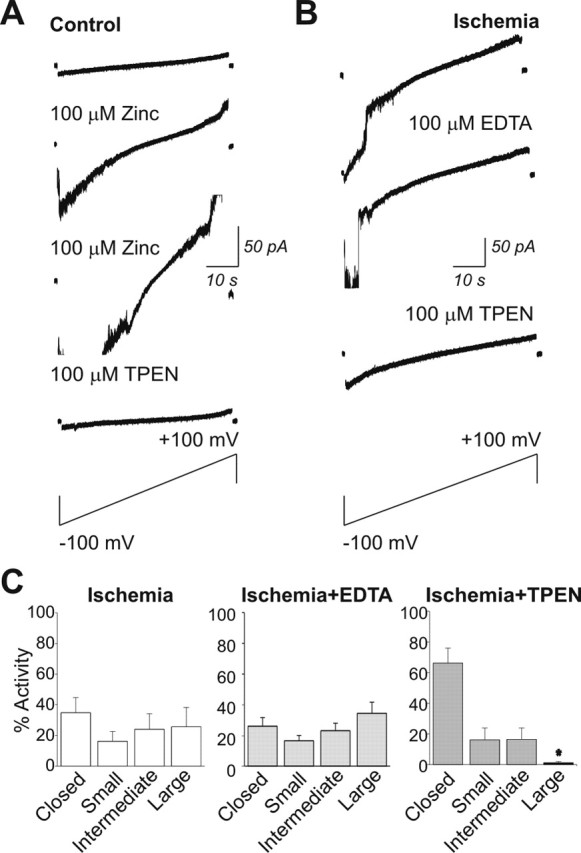
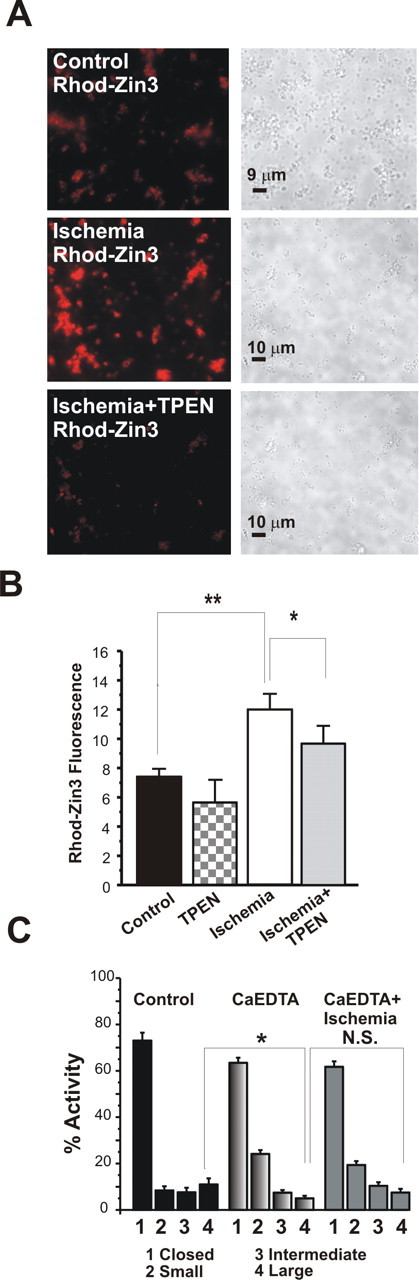
Transient global ischemia is a neuronal insult that induces delayed cell death. A hallmark event in the early post-ischemic period is enhanced permeability of mitochondrial membranes. The precise mechanisms by which mitochondrial function is disrupted are, as yet, unclear. Here we show that global ischemia promotes alterations in mitochondrial membrane contact points, a rise in intramitochondrial Zn2+, and activation of large, multi-conductance channels in mitochondrial outer membranes by 1 h after insult. Mitochondrial channel activity was associated with enhanced protease activity and proteolytic cleavage of BCL-xL to generate its pro-death counterpart, deltaN-BCL-xL. The findings implicate deltaN-BCL-xL in large, multi-conductance channel activity. Consistent with this, large channel activity was mimicked by introduction of recombinant deltaN-BCL-xL to control mitochondria and blocked by introduction of a functional BCL-xL antibody to post-ischemic mitochondria via the patch pipette. Channel activity was also inhibited by nicotinamide adenine dinucleotide, indicative of a role for the voltage-dependent anion channel (VDAC) of the outer mitochondrial membrane. In vivo administration of the membrane-impermeant Zn2+ chelator CaEDTA before ischemia or in vitro application of the membrane-permeant Zn2+ chelator tetrakis-(2-pyridylmethyl) ethylenediamine attenuated channel activity, suggesting a requirement for Zn2+. These findings reveal a novel mechanism by which ischemic insults disrupt the functional integrity of the outer mitochondrial membrane and implicate deltaN-BCL-xL and VDAC in the large, Zn2+-dependent mitochondrial channels observed in post-ischemic hippocampal mitochondria.
Figures
References
-
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ (2000). Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem 75:1878–1888. - PubMed
-
- Arslan P, Di Virgilio F, Beltrame M, Tsien RY, Pozzan T (1985). Cytosolic Ca2+ homeostasis in Ehrlich and Yoshida carcinomas. A new, membrane-permeant chelator of heavy metals reveals that these ascites tumor cell lines have normal cytosolic free Ca2+ J Biol Chem 260:2719–2727. - PubMed
-
- Assaf SY, Chung SH (1984). Release of endogenous Zn2+ from brain tissue during activity. Nature 308:734–736. - PubMed
-
- Banasiak KJ, Xia Y, Haddad GG (2000). Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol 62:215–249. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials