

Inducible proteopathies
- PMID: 16806508
- PMCID: PMC10725716
- DOI: 10.1016/j.tins.2006.06.010
Inducible proteopathies
Abstract
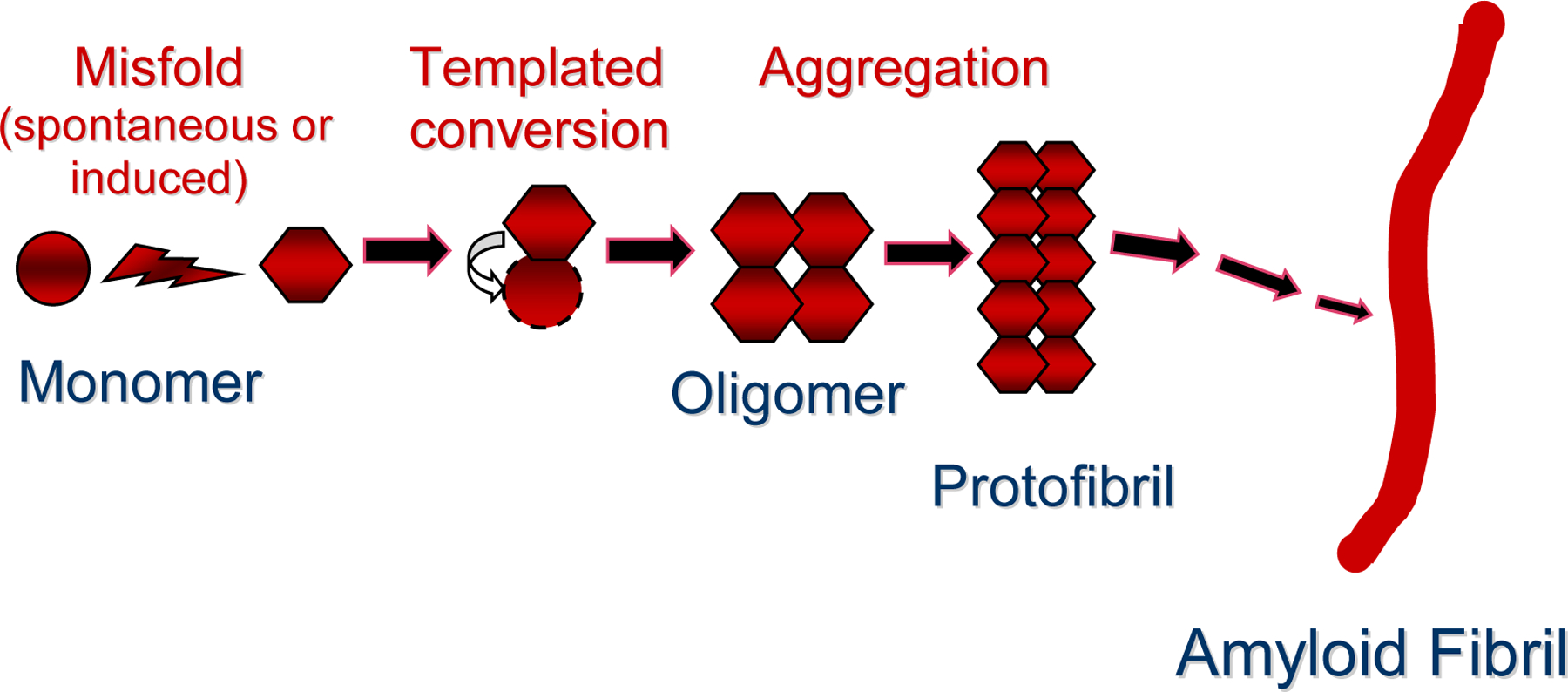
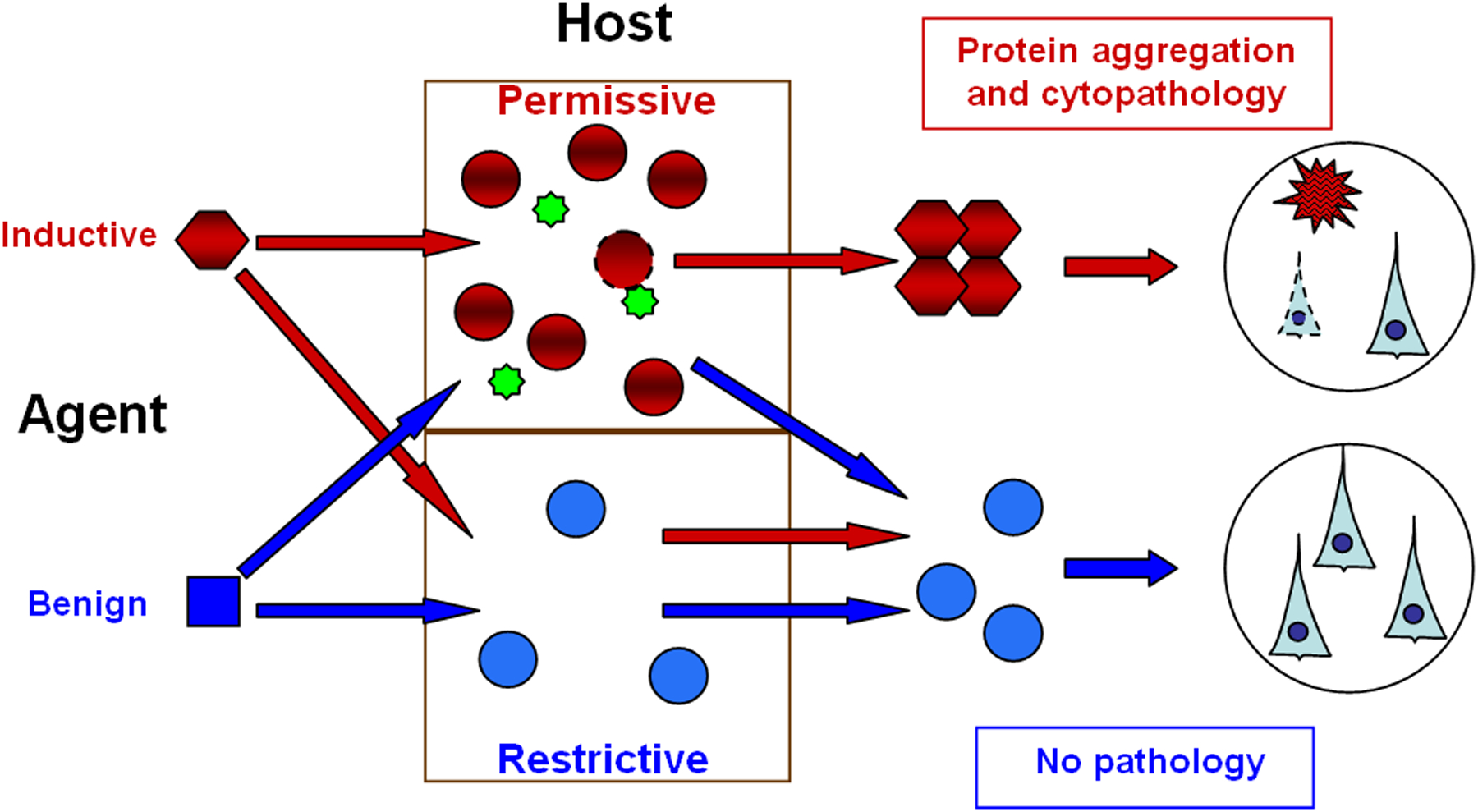
Numerous degenerative diseases are characterized by the aberrant polymerization and accumulation of specific proteins. These proteopathies include neurological disorders such as Alzheimer's disease, Parkinson's disease, Huntington's disease and the prion diseases, in addition to diverse systemic disorders, particularly the amyloidoses. The prion diseases have been shown to be transmissible by an alternative conformation of the normal cellular prion protein. Other proteopathies have been thought to be non-transmissible, but there is growing evidence that some systemic and cerebral amyloidoses can be induced by exposure of susceptible hosts to cognate molecular templates. As we review here, the mechanistic similarities among these diseases provide unprecedented opportunities for elucidating the induction of protein misfolding and assembly in vivo, and for developing an integrated therapeutic approach to degenerative proteopathies.
Figures
References
-
- Walker LC and LeVine H (2000) The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly. Mol Neurobiol 21, 83–95 - PubMed
-
- Prusiner SB (2001) Shattuck lecture--neurodegenerative diseases and prions. N Engl J Med 344, 1516–1526 - PubMed
-
- Carrell RW and Lomas DA (2002) Alpha1-antitrypsin deficiency--a model for conformational diseases. N Engl J Med 346, 45–53 - PubMed
-
- Hardy J and Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 - PubMed
-
- Johnson RT (2005) Prion diseases. Lancet Neurol 4, 635–642 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
