

PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons
- PMID: 16809306
- PMCID: PMC1489062
- DOI: 10.1128/JVI.00489-06
PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons
Abstract
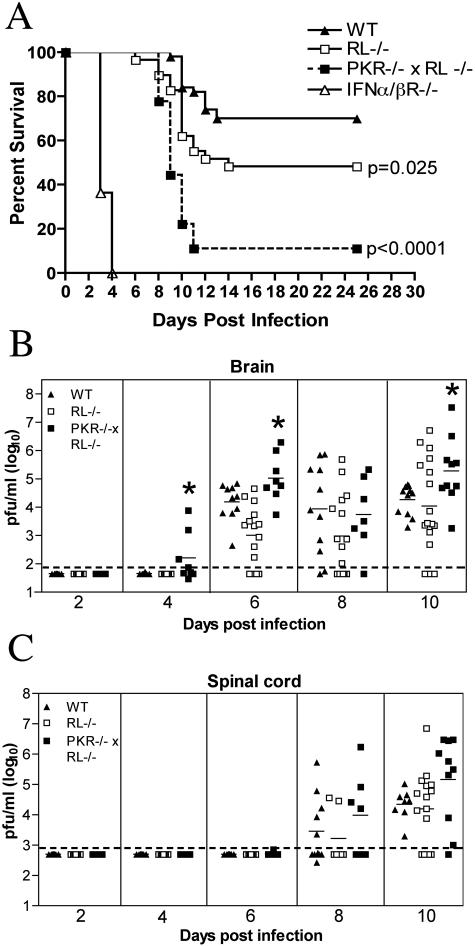
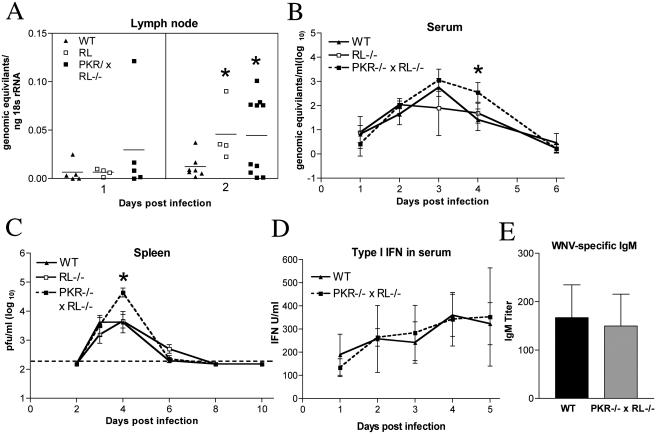
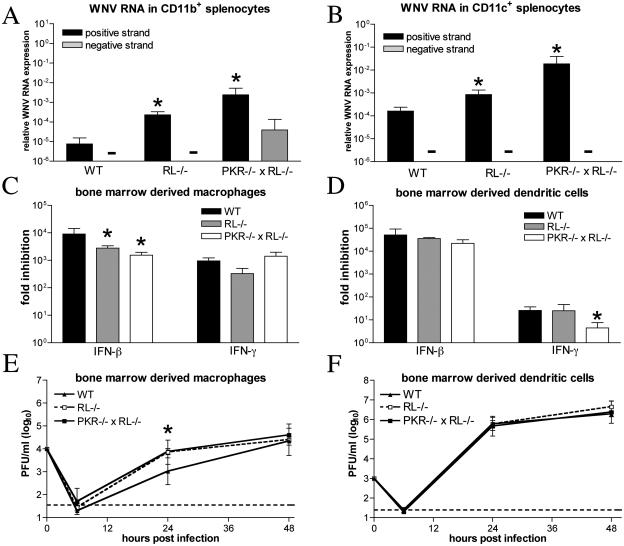
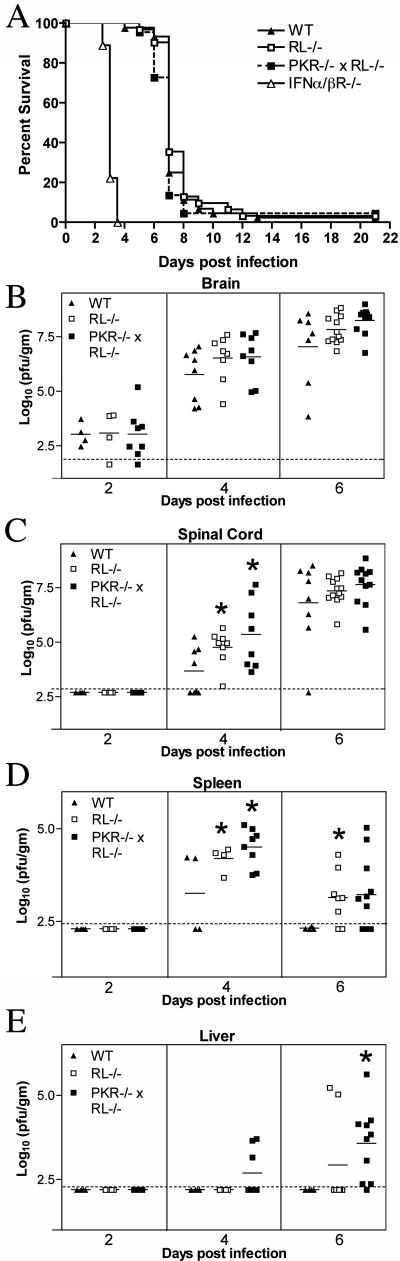
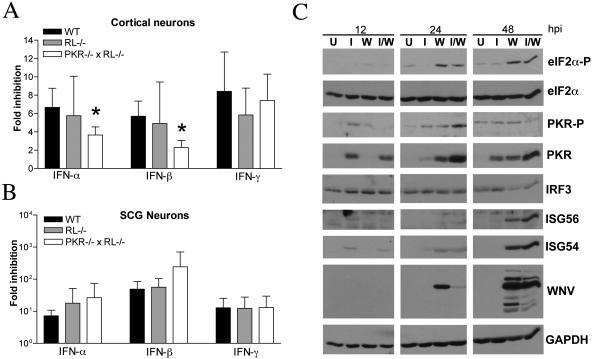
West Nile virus (WNV) is a neurotropic, mosquito-borne flavivirus that can cause lethal meningoencephalitis. Type I interferon (IFN) plays a critical role in controlling WNV replication, spread, and tropism. In this study, we begin to examine the effector mechanisms by which type I IFN inhibits WNV infection. Mice lacking both the interferon-induced, double-stranded-RNA-activated protein kinase (PKR) and the endoribonuclease of the 2',5'-oligoadenylate synthetase-RNase L system (PKR(-/-) x RL(-/-)) were highly susceptible to subcutaneous WNV infection, with a 90% mortality rate compared to the 30% mortality rate observed in congenic wild-type mice. PKR(-/-) x RL(-/-) mice had increased viral loads in their draining lymph nodes, sera, and spleens, which led to early viral entry into the central nervous system (CNS) and higher viral burden in neuronal tissues. Although mice lacking RNase L showed a higher CNS viral burden and an increased mortality, they were less susceptible than the PKR(-/-) x RL(-/-) mice; thus, we also infer an antiviral role for PKR in the control of WNV infection. Notably, a deficiency in both PKR and RNase L resulted in a decreased ability of type I IFN to inhibit WNV in primary macrophages and cortical neurons. In contrast, the peripheral neurons of the superior cervical ganglia of PKR(-/-) x RL(-/-) mice showed no deficiency in the IFN-mediated inhibition of WNV. Our data suggest that PKR and RNase L contribute to IFN-mediated protection in a cell-restricted manner and control WNV infection in peripheral tissues and some neuronal subtypes.
Figures
References
-
- Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732-738. - PubMed
-
- Al-khatib, K., B. R. Williams, R. H. Silverman, W. Halford, and D. J. Carr. 2003. The murine double-stranded RNA-dependent protein kinase PKR and the murine 2′,5′-oligoadenylate synthetase-dependent RNase L are required for IFN-beta-mediated resistance against herpes simplex virus type 1 in primary trigeminal ganglion culture. Virology 313:126-135. - PubMed
-
- Asnis, D. S., R. Conetta, A. A. Teixeira, G. Waldman, and B. A. Sampson. 2000. The West Nile virus outbreak of 1999 in New York: the Flushing Hospital experience. Clin. Infect. Dis. 30:413-418. - PubMed
-
- Austin, B. A., C. James, R. H. Silverman, and D. J. Carr. 2005. Critical role for the oligoadenylate synthetase/RNase L pathway in response to IFN-beta during acute ocular herpes simplex virus type 1 infection. J. Immunol. 175:1100-1106. - PubMed
-
- Balachandran, S., P. C. Roberts, L. E. Brown, H. Truong, A. K. Pattnaik, D. R. Archer, and G. N. Barber. 2000. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13:129-141. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
