

Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1
- PMID: 16809319
- PMCID: PMC1489027
- DOI: 10.1128/JVI.00509-06
Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1
Abstract
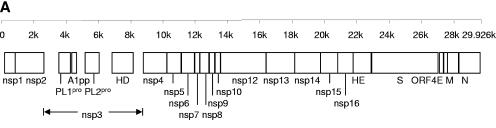
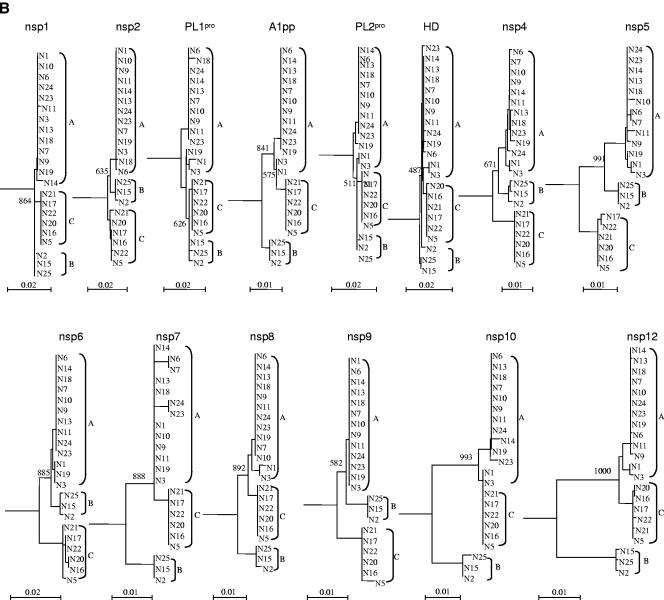
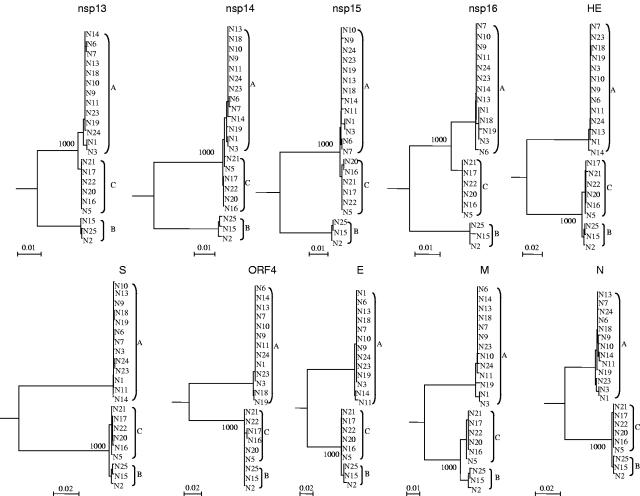
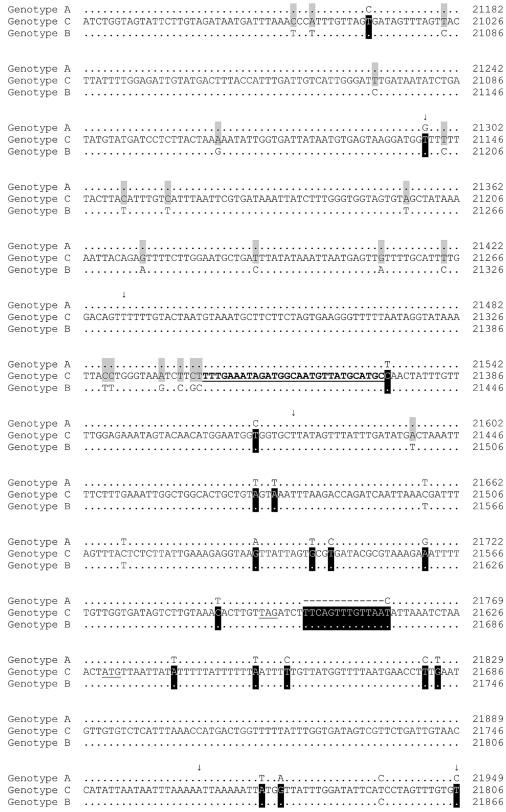
We sequenced and compared the complete genomes of 22 strains of coronavirus HKU1 (CoV HKU1) obtained from nasopharyngeal aspirates of patients with respiratory tract infections over a 2-year period. Phylogenetic analysis of 24 putative proteins and polypeptides showed that the 22 CoV HKU1 strains fell into three clusters (genotype A, 13 strains; genotype B, 3 strains and genotype C, 6 strains). However, different phylogenetic relationships among the three clusters were observed in different regions of their genomes. From nsp4 to nsp6, the genotype A strains were clustered with the genotype B strains. For nsp7 and nsp8 and from nsp10 to nsp16, the genotype A strains were clustered with the genotype C strains. From hemagglutinin esterase (HE) to nucleocapsid (N), the genotype B strains were clustered closely with the genotype C strains. Bootscan analysis showed possible recombination between genotypes B and C from nucleotide positions 11,500 to 13,000, corresponding to the nsp6-nsp7 junction, giving rise to genotype A, and between genotypes A and B from nucleotide positions 21,500 to 22,500, corresponding to the nsp16-HE junction, giving rise to genotype C. Multiple alignments further narrowed the sites of crossover to a 143-bp region between nucleotide positions 11,750 and 11,892 and a 29-bp region between nucleotide positions 21,502 and 21,530. Genome analysis also revealed various numbers of tandem copies of a perfect 30-base acidic tandem repeat (ATR) which encodes NDDEDVVTGD and various numbers and sequences of imperfect repeats in the N terminus of nsp3 inside the acidic domain upstream of papain-like protease 1 among the 22 genomes. All 10 CoV HKU1 strains with incomplete imperfect repeats (1.4 and 4.4) belonged to genotype A. The present study represents the first evidence for natural recombination in coronavirus associated with human infection. Analysis of a single gene is not sufficient for the genotyping of CoV HKU1 strains but requires amplification and sequencing of at least two gene loci, one from nsp10 to nsp16 (e.g., pol or helicase) and another from HE to N (e.g., spike or N). Further studies will delineate whether the ATR is useful for the molecular typing of CoV HKU1.
Figures


References
-
- Bierne, H., M. Seigneur, S. D. Ehrlich, and B. Michel. 1997. uvrD mutations enhance tandem repeat deletion in the Escherichia coli chromosome via SOS induction of the RecF recombination pathway. Mol. Microbiol. 26:557-567. - PubMed
-
- Copper, P. D., A. Steiner-Pryor, P. D. Scotti, and D. Delong. 1974. On the nature of poliovirus genetic recombinants. J. Gen. Virol. 23:41-49. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
