

Sequence of events in folding mechanism: beyond the Gō model
- PMID: 16815916
- PMCID: PMC2242563
- DOI: 10.1110/ps.052056006
Sequence of events in folding mechanism: beyond the Gō model
Abstract
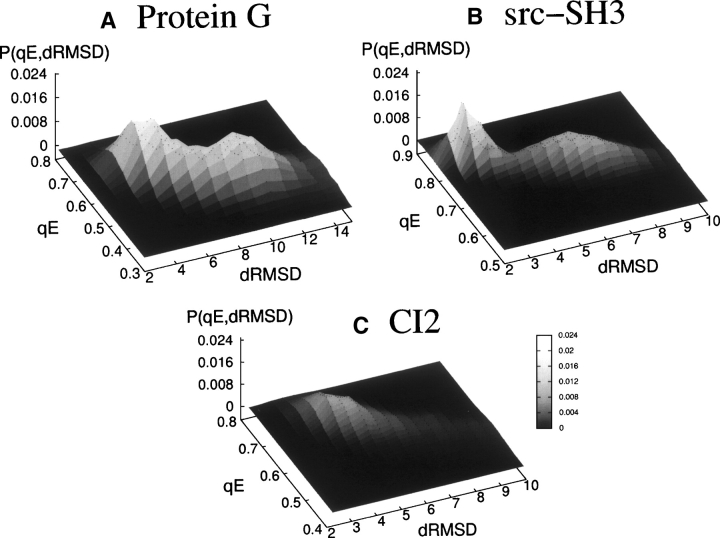
Simplified Gō models, where only native contacts interact favorably, have proven useful to characterize some aspects of the folding of small proteins. The success of these models is limited by the fact that all residues interact in the same way so that the folding features of a protein are determined only by the geometry of its native conformation. We present an extended version of a Calpha-based Gō model where different residues interact with different energies. The model is used to calculate the thermodynamics of three small proteins (Protein G, Src-SH3, and CI2) and the effect of mutations (DeltaDeltaGU-N, DeltaDeltaGdouble dagger-N, DeltaDeltaGdouble dagger-U, and phi-values) on the wild-type sequence. The model allows us to investigate some of the most controversial areas in protein folding, such as its earliest stages and the nature of the unfolded state, subjects that have lately received particular attention.
Figures










References
-
- Abkevich V.I., Gutin A.M., Shakhnovich E.I. 1994. Specific nucleus as the transition state for protein folding. Biochemistry 33: 10026–10031. - PubMed
-
- Alexander P., Fahnestock S., Lee T., Orban J., Bryan P. 1992. Thermodynamic analysis of the folding of the streptococcal Protein G IgG-binding domains B1 and B2. Biochememistry 31: 3597–3603. - PubMed
-
- Baldwin R.L. and Rose G.D. 1999. Is protein folding hierarchic? Trends Biochem. Sci. 24: 26–33. - PubMed
-
- Berendsen H.J.C., van der Spoel D., van Drunen R. 1995. GROMACS: A message–passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91: 43–55.
-
- Blanco F.G., Rivas G., Serrano L. 1994. Folding of protein G B1 domain studied by conformational characterization of fragments comprising its secondary structure. Eur. J. Biochem. 230: 634–649. - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Miscellaneous
