

X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production
- PMID: 16818673
- PMCID: PMC2118353
- DOI: 10.1084/jem.20060085
X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production
Abstract
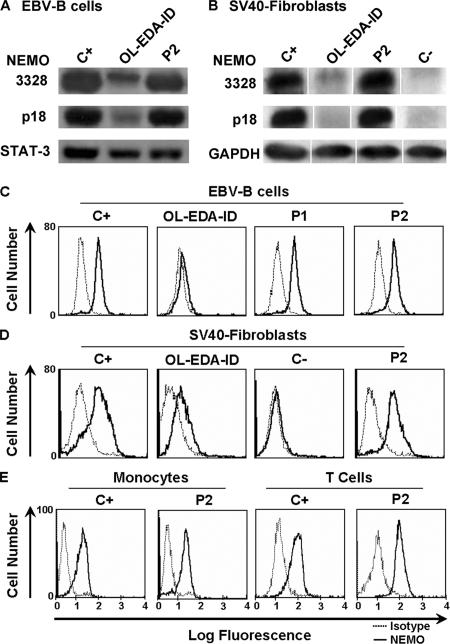
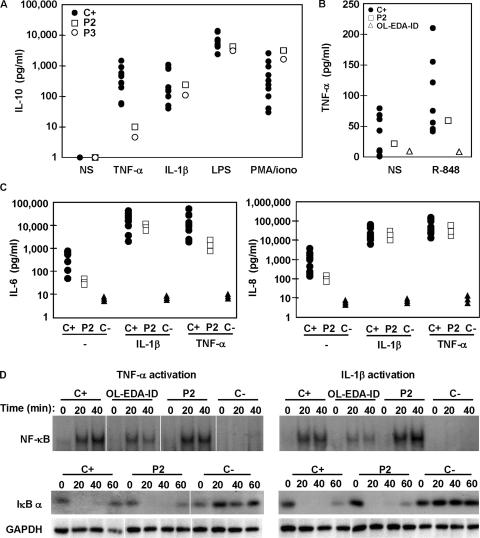
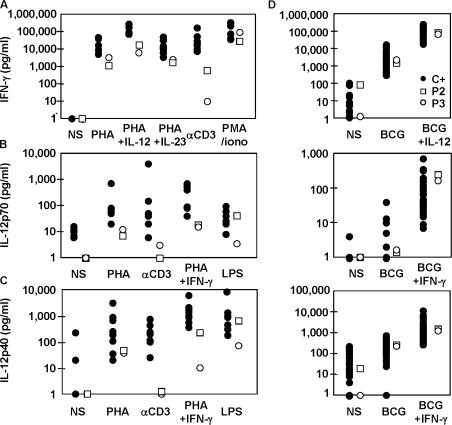
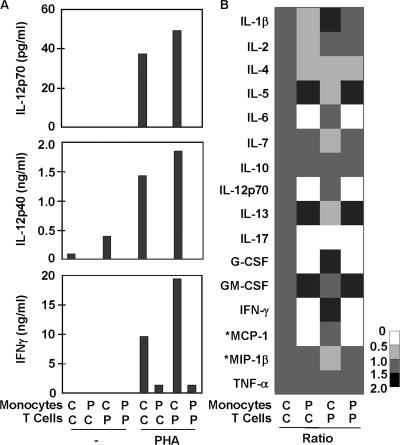
Germline mutations in five autosomal genes involved in interleukin (IL)-12-dependent, interferon (IFN)-gamma-mediated immunity cause Mendelian susceptibility to mycobacterial diseases (MSMD). The molecular basis of X-linked recessive (XR)-MSMD remains unknown. We report here mutations in the leucine zipper (LZ) domain of the NF-kappaB essential modulator (NEMO) gene in three unrelated kindreds with XR-MSMD. The mutant proteins were produced in normal amounts in blood and fibroblastic cells. However, the patients' monocytes presented an intrinsic defect in T cell-dependent IL-12 production, resulting in defective IFN-gamma secretion by T cells. IL-12 production was also impaired as the result of a specific defect in NEMO- and NF-kappaB/c-Rel-mediated CD40 signaling after the stimulation of monocytes and dendritic cells by CD40L-expressing T cells and fibroblasts, respectively. However, the CD40-dependent up-regulation of costimulatory molecules of dendritic cells and the proliferation and immunoglobulin class switch of B cells were normal. Moreover, the patients' blood and fibroblastic cells responded to other NF-kappaB activators, such as tumor necrosis factor-alpha, IL-1beta, and lipopolysaccharide. These two mutations in the NEMO LZ domain provide the first genetic etiology of XR-MSMD. They also demonstrate the importance of the T cell- and CD40L-triggered, CD40-, and NEMO/NF-kappaB/c-Rel-mediated induction of IL-12 by monocyte-derived cells for protective immunity to mycobacteria in humans.
Figures



References
-
- Holland, S.M., E.M. Eisenstein, D.B. Kuhns, M.L. Turner, T.A. Fleisher, W. Strober, and J.I. Gallin. 1994. Treatment of refractory disseminated nontuberculous mycobacterial infection with interferon γ. A preliminary report. N. Engl. J. Med. 330:1348–1355. - PubMed
-
- Levin, M., M.J. Newport, S. D'Souza, P. Kalabalikis, I.N. Brown, H.M. Lenicker, P.V. Agius, E.G. Davies, A. Thrasher, N. Klein, and J. Blackwell. 1995. Familial disseminated atypical mycobacterial infection in childhood: a human mycobacterial susceptibility gene? Lancet. 345:79–83. - PubMed
-
- Casanova, J.L., E. Jouanguy, S. Lamhamedi, S. Blanche, and A. Fischer. 1995. Immunological conditions of children with BCG disseminated infection. Lancet. 346:581. - PubMed
-
- Casanova, J.L., and L. Abel. 2002. Genetic dissection of immunity to mycobacteria: the human model. Annu. Rev. Immunol. 20:581–620. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
