

PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins
- PMID: 16818720
- PMCID: PMC2064165
- DOI: 10.1083/jcb.200511045
PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins
Abstract
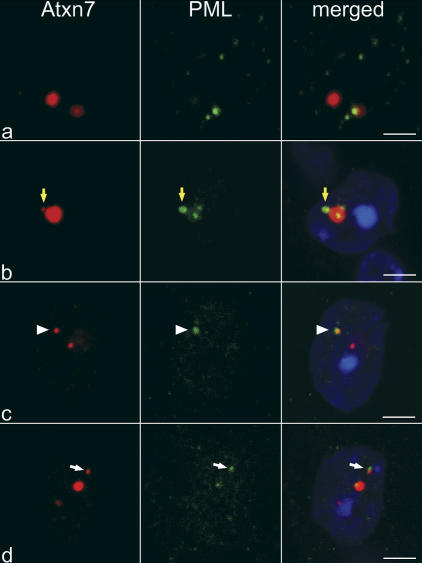
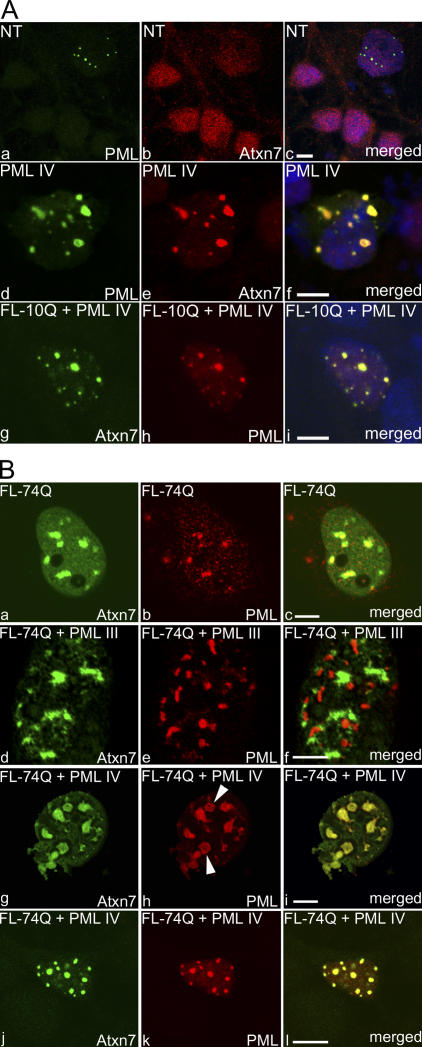
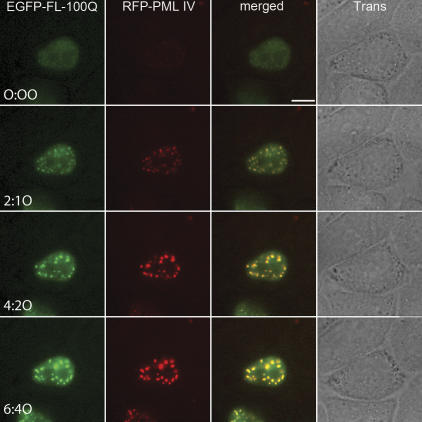
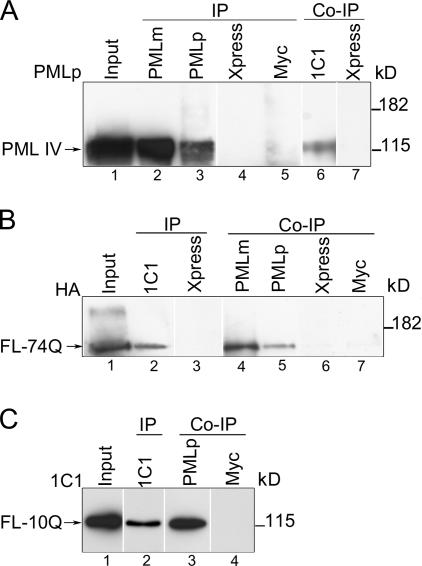
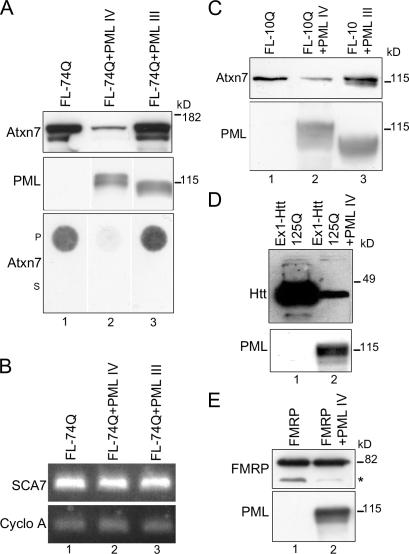
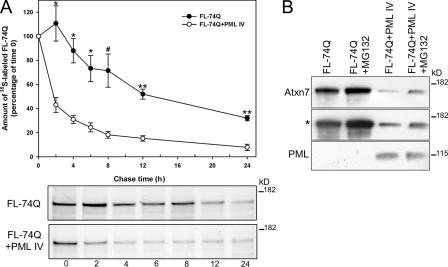
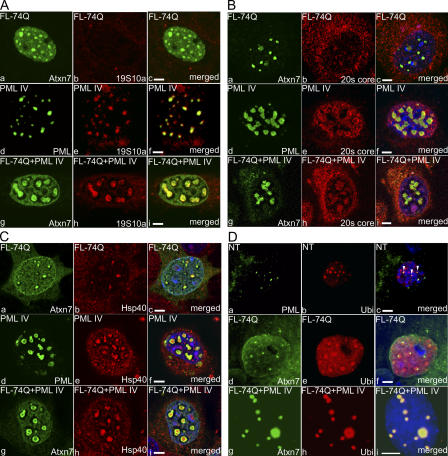
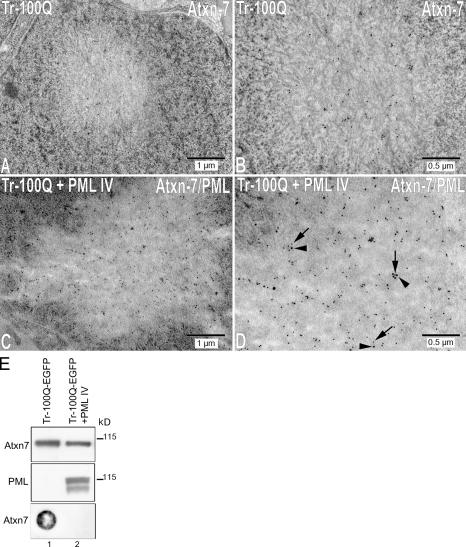
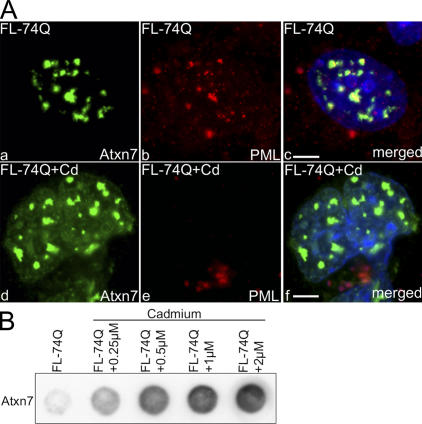
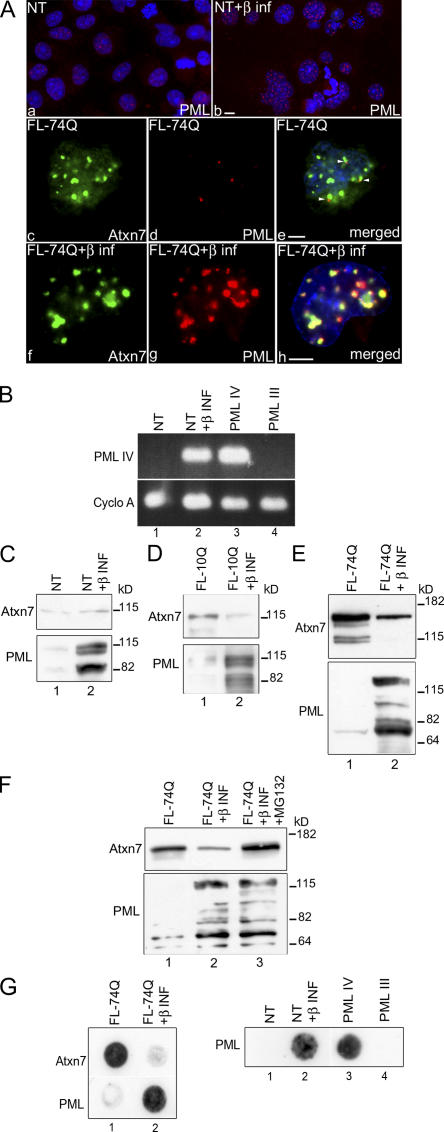
The pathogenesis of spinocerebellar ataxia type 7 and other neurodegenerative polyglutamine (polyQ) disorders correlates with the aberrant accumulation of toxic polyQ-expanded proteins in the nucleus. Promyelocytic leukemia protein (PML) nuclear bodies are often present in polyQ aggregates, but their relation to pathogenesis is unclear. We show that expression of PML isoform IV leads to the formation of distinct nuclear bodies enriched in components of the ubiquitin-proteasome system. These bodies recruit soluble mutant ataxin-7 and promote its degradation by proteasome-dependent proteolysis, thus preventing the aggregate formation. Inversely, disruption of the endogenous nuclear bodies with cadmium increases the nuclear accumulation and aggregation of mutant ataxin-7, demonstrating their role in ataxin-7 turnover. Interestingly, beta-interferon treatment, which induces the expression of endogenous PML IV, prevents the accumulation of transiently expressed mutant ataxin-7 without affecting the level of the endogenous wild-type protein. Therefore, clastosomes represent a potential therapeutic target for preventing polyQ disorders.
Figures
Similar articles
-
Promyelocytic leukemia protein is redistributed during the formation of intranuclear inclusions independent of polyglutamine expansion: an immunohistochemical study on Marinesco bodies.J Neuropathol Exp Neurol. 2002 Nov;61(11):984-91. doi: 10.1093/jnen/61.11.984. J Neuropathol Exp Neurol. 2002. PMID: 12430715
-
Transcriptional repression and cell death induced by nuclear aggregates of non-polyglutamine protein.Neurobiol Dis. 2005 Dec;20(3):656-65. doi: 10.1016/j.nbd.2005.05.015. Epub 2005 Jun 16. Neurobiol Dis. 2005. PMID: 15964198 Free PMC article.
-
H1152 promotes the degradation of polyglutamine-expanded ataxin-3 or ataxin-7 independently of its ROCK-inhibiting effect and ameliorates mutant ataxin-3-induced neurodegeneration in the SCA3 transgenic mouse.Neuropharmacology. 2013 Jul;70:1-11. doi: 10.1016/j.neuropharm.2013.01.006. Epub 2013 Jan 21. Neuropharmacology. 2013. PMID: 23347954
-
Progress in pathogenesis studies of spinocerebellar ataxia type 1.Philos Trans R Soc Lond B Biol Sci. 1999 Jun 29;354(1386):1079-81. doi: 10.1098/rstb.1999.0462. Philos Trans R Soc Lond B Biol Sci. 1999. PMID: 10434309 Free PMC article. Review.
-
Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies.Mol Cell Biol. 2002 Aug;22(15):5259-69. doi: 10.1128/MCB.22.15.5259-5269.2002. Mol Cell Biol. 2002. PMID: 12101223 Free PMC article. Review. No abstract available.
Cited by
-
Assays for the Degradation of Misfolded Proteins in Cells.J Vis Exp. 2016 Aug 28;(114):54266. doi: 10.3791/54266. J Vis Exp. 2016. PMID: 27684355 Free PMC article.
-
Running 'LAPS' Around nLD: Nuclear Lipid Droplet Form and Function.Front Cell Dev Biol. 2022 Feb 1;10:837406. doi: 10.3389/fcell.2022.837406. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35178392 Free PMC article. Review.
-
PML: Regulation and multifaceted function beyond tumor suppression.Cell Biosci. 2018 Jan 25;8:5. doi: 10.1186/s13578-018-0204-8. eCollection 2018. Cell Biosci. 2018. PMID: 29416846 Free PMC article. Review.
-
Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype.PLoS One. 2009 Sep 30;4(9):e7232. doi: 10.1371/journal.pone.0007232. PLoS One. 2009. PMID: 19789634 Free PMC article.
-
Orphan nuclear bodies.Cold Spring Harb Perspect Biol. 2010 Sep;2(9):a000703. doi: 10.1101/cshperspect.a000703. Epub 2010 Jul 7. Cold Spring Harb Perspect Biol. 2010. PMID: 20610547 Free PMC article. Review.
References
-
- Beech, S.J., K.J. Lethbridge, N. Killick, N. McGlincy, and K.N. Leppard. 2005. Isoforms of the promyelocytic leukemia protein differ in their effects on ND10 organization. Exp. Cell Res. 307:109–117. - PubMed
-
- Cancel, G., C. Duyckaerts, M. Holmberg, C. Zander, G. Yvert, A.S. Lebre, M. Ruberg, B. Faucheux, Y. Agid, E. Hirsch, and A. Brice. 2000. Distribution of ataxin-7 in normal human brain and retina. Brain. 123:2519–2530. - PubMed
-
- Chai, Y., S.L. Koppenhafer, S.J. Shoesmith, M.K. Perez, and H.L. Paulson. 1999. Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum. Mol. Genet. 8:673–682. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
