

Central insulin action in energy and glucose homeostasis
- PMID: 16823473
- PMCID: PMC1483153
- DOI: 10.1172/JCI29063
Central insulin action in energy and glucose homeostasis
Abstract
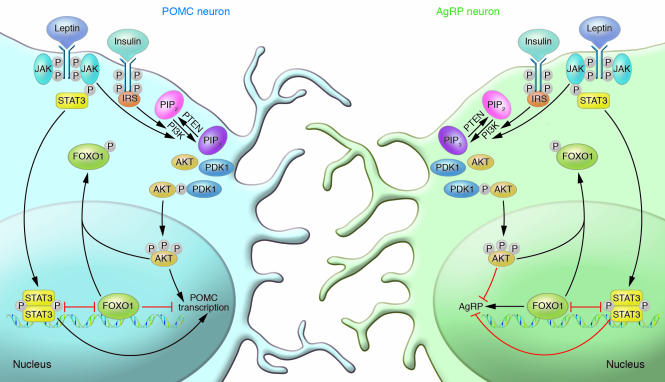
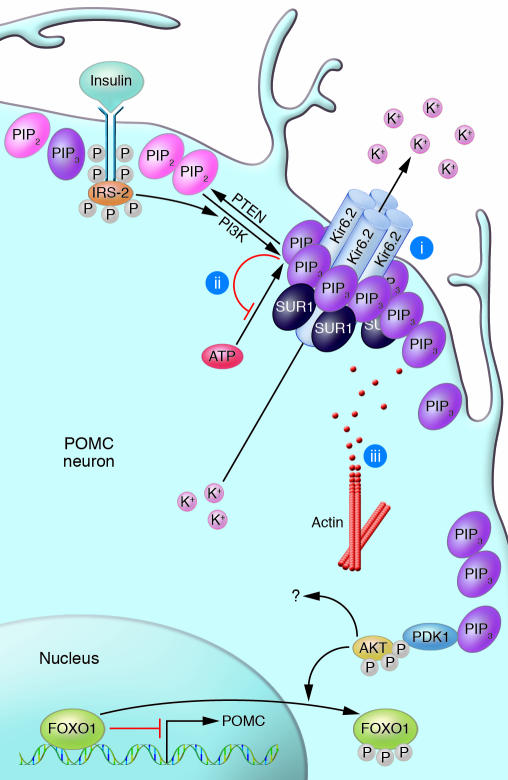
Insulin has pleiotropic biological effects in virtually all tissues. However, the relevance of insulin signaling in peripheral tissues has been studied far more extensively than its role in the brain. An evolving body of evidence indicates that in the brain, insulin is involved in multiple regulatory mechanisms including neuronal survival, learning, and memory, as well as in regulation of energy homeostasis and reproductive endocrinology. Here we review insulin's role as a central homeostatic signal with regard to energy and glucose homeostasis and discuss the mechanisms by which insulin communicates information about the body's energy status to the brain. Particular emphasis is placed on the controversial current debate about the similarities and differences between hypothalamic insulin and leptin signaling at the molecular level.
Figures
References
-
- Bernard C.1855. . Leçons de physiologie expérimentale appliquée à la médecine faites au Collège de France. Baillère et Fils.Paris, France.296–313.
-
- Rulifson E.J., Kim S.K., Nusse R. Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science. 2002;296:1118–1120. - PubMed
-
- Wolkow C.A., Kimura K.D., Lee M.S., Ruvkun G. Regulation of C. elegans life-span by insulinlike signaling in the nervous system. Science. 2000;290:147–150. - PubMed
