

Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy
- PMID: 16823493
- PMCID: PMC1483165
- DOI: 10.1172/JCI27751
Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy
Abstract
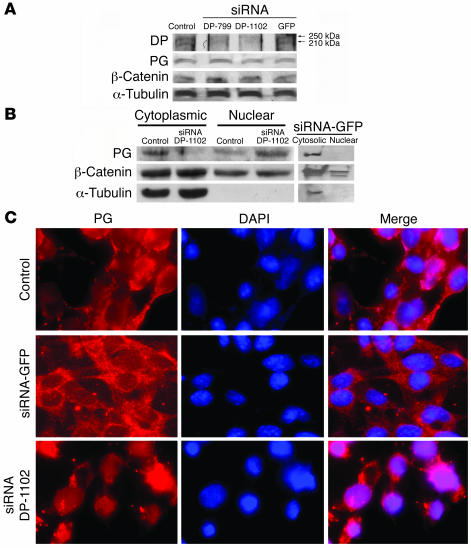
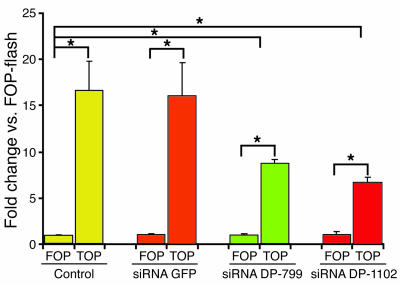
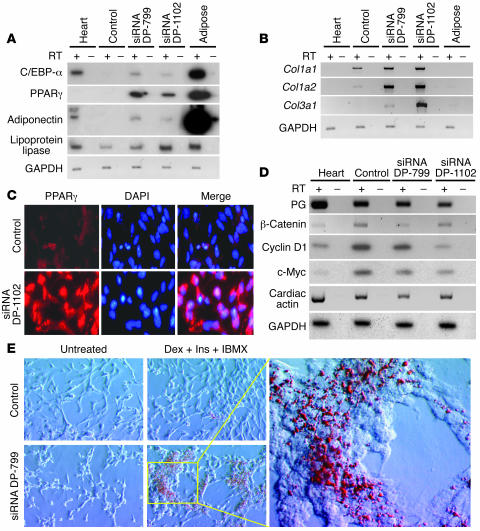
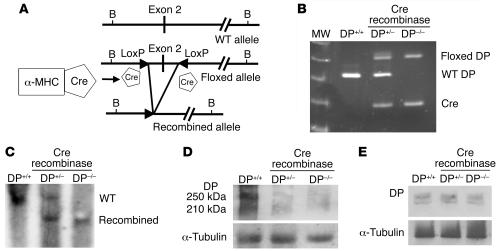
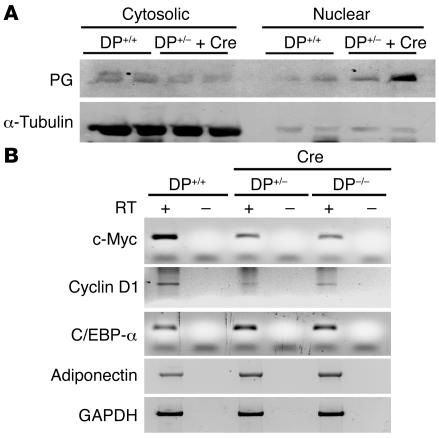
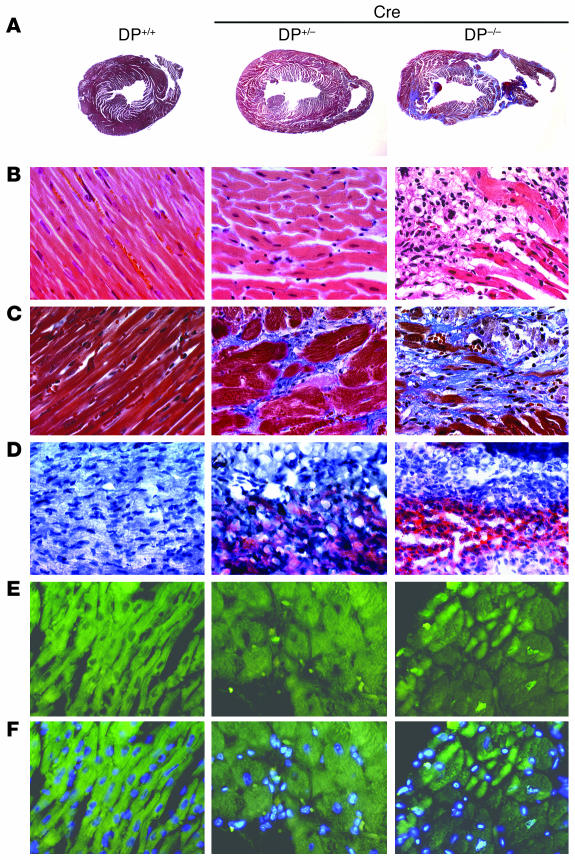
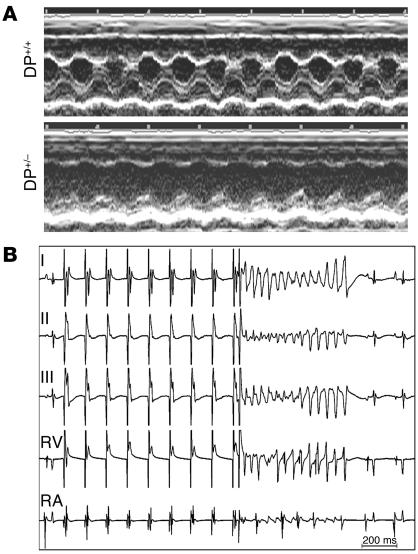
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVC) is a genetic disease caused by mutations in desmosomal proteins. The phenotypic hallmark of ARVC is fibroadipocytic replacement of cardiac myocytes, which is a unique phenotype with a yet-to-be-defined molecular mechanism. We established atrial myocyte cell lines expressing siRNA against desmoplakin (DP), responsible for human ARVC. We show suppression of DP expression leads to nuclear localization of the desmosomal protein plakoglobin and a 2-fold reduction in canonical Wnt/beta-catenin signaling through Tcf/Lef1 transcription factors. The ensuing phenotype is increased expression of adipogenic and fibrogenic genes and accumulation of fat droplets. We further show that cardiac-restricted deletion of Dsp, encoding DP, impairs cardiac morphogenesis and leads to high embryonic lethality in the homozygous state. Heterozygous DP-deficient mice exhibited excess adipocytes and fibrosis in the myocardium, increased myocyte apoptosis, cardiac dysfunction, and ventricular arrhythmias, thus recapitulating the phenotype of human ARVC. We believe our results provide for a novel molecular mechanism for the pathogenesis of ARVC and establish cardiac-restricted DP-deficient mice as a model for human ARVC. These findings could provide for the opportunity to identify new diagnostic markers and therapeutic targets in patients with ARVC.
Figures
Comment in
-
Arrhythmogenic right ventricular cardiomyopathy: moving toward mechanism.J Clin Invest. 2006 Jul;116(7):1825-8. doi: 10.1172/JCI29174. J Clin Invest. 2006. PMID: 16823481 Free PMC article.
References
-
- Corrado D., et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J. Am. Coll. Cardiol. 1997;30:1512–1520. - PubMed
-
- Sen-Chowdhry S., Syrris P., McKenna W.J. Genetics of right ventricular cardiomyopathy. . J. Cardiovasc. Electrophysiol. 2005;16:927–935. - PubMed
-
- Maron B.J., et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199–204. - PubMed
-
- Tabib A., et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation. 2003;108:3000–3005. - PubMed
-
- Kaplan S.R., et al. Structural and molecular pathology of the heart in Carvajal syndrome. . Cardiovasc. Pathol. 2004;13:26–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
