

Automated SNP detection from a large collection of white spruce expressed sequences: contributing factors and approaches for the categorization of SNPs
- PMID: 16824208
- PMCID: PMC1557672
- DOI: 10.1186/1471-2164-7-174
Automated SNP detection from a large collection of white spruce expressed sequences: contributing factors and approaches for the categorization of SNPs
Abstract
Background: High-throughput genotyping technologies represent a highly efficient way to accelerate genetic mapping and enable association studies. As a first step toward this goal, we aimed to develop a resource of candidate Single Nucleotide Polymorphisms (SNP) in white spruce (Picea glauca [Moench] Voss), a softwood tree of major economic importance.
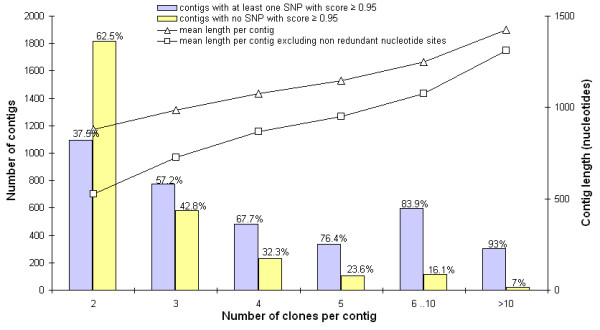
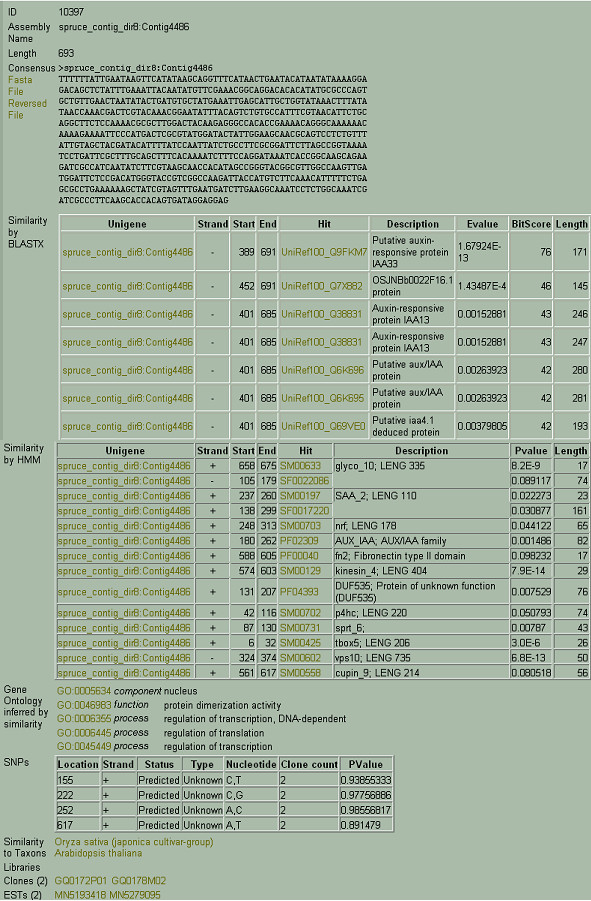
Results: A white spruce SNP resource encompassing 12,264 SNPs was constructed from a set of 6,459 contigs derived from Expressed Sequence Tags (EST) and by using the bayesian-based statistical software PolyBayes. Several parameters influencing the SNP prediction were analysed including the a priori expected polymorphism, the probability score (PSNP), and the contig depth and length. SNP detection in 3' and 5' reads from the same clones revealed a level of inconsistency between overlapping sequences as low as 1%. A subset of 245 predicted SNPs were verified through the independent resequencing of genomic DNA of a genotype also used to prepare cDNA libraries. The validation rate reached a maximum of 85% for SNPs predicted with either PSNP > or = 0.95 or > or = 0.99. A total of 9,310 SNPs were detected by using PSNP > or = 0.95 as a criterion. The SNPs were distributed among 3,590 contigs encompassing an array of broad functional categories, with an overall frequency of 1 SNP per 700 nucleotide sites. Experimental and statistical approaches were used to evaluate the proportion of paralogous SNPs, with estimates in the range of 8 to 12%. The 3,789 coding SNPs identified through coding region annotation and ORF prediction, were distributed into 39% nonsynonymous and 61% synonymous substitutions. Overall, there were 0.9 SNP per 1,000 nonsynonymous sites and 5.2 SNPs per 1,000 synonymous sites, for a genome-wide nonsynonymous to synonymous substitution rate ratio (Ka/Ks) of 0.17.
Conclusion: We integrated the SNP data in the ForestTreeDB database along with functional annotations to provide a tool facilitating the choice of candidate genes for mapping purposes or association studies.
Figures


Similar articles
-
The landscape of nucleotide polymorphism among 13,500 genes of the conifer picea glauca, relationships with functions, and comparison with medicago truncatula.Genome Biol Evol. 2013;5(10):1910-25. doi: 10.1093/gbe/evt143. Genome Biol Evol. 2013. PMID: 24065735 Free PMC article.
-
Generation, annotation, analysis and database integration of 16,500 white spruce EST clusters.BMC Genomics. 2005 Oct 19;6:144. doi: 10.1186/1471-2164-6-144. BMC Genomics. 2005. PMID: 16236172 Free PMC article.
-
Generation, functional annotation and comparative analysis of black spruce (Picea mariana) ESTs: an important conifer genomic resource.BMC Genomics. 2013 Oct 11;14:702. doi: 10.1186/1471-2164-14-702. BMC Genomics. 2013. PMID: 24119028 Free PMC article.
-
Tools, resources and databases for SNPs and indels in sequences: a review.Int J Bioinform Res Appl. 2014;10(3):264-96. doi: 10.1504/IJBRA.2014.060762. Int J Bioinform Res Appl. 2014. PMID: 24794070 Review.
-
Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: current and developing technologies.Mutat Res. 2008 Jul-Aug;659(1-2):147-57. doi: 10.1016/j.mrrev.2008.05.001. Epub 2008 May 4. Mutat Res. 2008. PMID: 18565787 Free PMC article. Review.
Cited by
-
An efficient method for developing SNP markers based on EST data combined with high resolution melting (HRM) analysis.BMC Res Notes. 2010 Mar 2;3:51. doi: 10.1186/1756-0500-3-51. BMC Res Notes. 2010. PMID: 20193087 Free PMC article.
-
The heterogeneous levels of linkage disequilibrium in white spruce genes and comparative analysis with other conifers.Heredity (Edinb). 2012 Mar;108(3):273-84. doi: 10.1038/hdy.2011.72. Epub 2011 Sep 7. Heredity (Edinb). 2012. PMID: 21897435 Free PMC article.
-
The landscape of nucleotide polymorphism among 13,500 genes of the conifer picea glauca, relationships with functions, and comparison with medicago truncatula.Genome Biol Evol. 2013;5(10):1910-25. doi: 10.1093/gbe/evt143. Genome Biol Evol. 2013. PMID: 24065735 Free PMC article.
-
A catalogue of putative unique transcripts from Douglas-fir (Pseudotsuga menziesii) based on 454 transcriptome sequencing of genetically diverse, drought stressed seedlings.BMC Genomics. 2012 Nov 28;13:673. doi: 10.1186/1471-2164-13-673. BMC Genomics. 2012. PMID: 23190494 Free PMC article.
-
Towards decoding the conifer giga-genome.Plant Mol Biol. 2012 Dec;80(6):555-69. doi: 10.1007/s11103-012-9961-7. Epub 2012 Sep 9. Plant Mol Biol. 2012. PMID: 22960864 Review.
References
-
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, Hunt SE, Cole CG, Coggill PC, Rice CM, Ning Z, Rogers J, Bentley DR, Kwok PY, Mardis ER, Yeh RT, Schultz B, Cook L, Davenport R, Dante M, Fulton L, Hillier L, Waterston RH, McPherson JD, Gilman B, Schaffner S, Van Etten WJ, Reich D, Higgins J, Daly MJ, Blumenstiel B, Baldwin J, Stange-Thomann N, Zody MC, Linton L, Lander ES, Altshuler D. International SNP Map Working Group. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. - PubMed
-
- Matise TC, Sachidanandam R, Clark AG, Kruglyak L, Wijsman E, Kakol J, Buyske S, Chui B, Cohen P, de Toma C, Ehm M, Glanowski S, He C, Heil J, Markianos K, McMullen I, Pericak-Vance MA, Silbergleit A, Stein L, Wagner M, Wilson AF, Winick JD, Winn-Deen ES, Yamashiro CT, Cann HM, Lai E, Holden AL. A 3.9-centimorgan-resolution human single-nucleotide polymorphism linkage map and screening set. Am J Hum Genet. 2003;73:271–284. - PMC - PubMed
-
- The Arabidopsis Information Resource http://www.arabidopis.org/
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
