

Genomic signatures of positive selection in humans and the limits of outlier approaches
- PMID: 16825663
- PMCID: PMC1524870
- DOI: 10.1101/gr.5157306
Genomic signatures of positive selection in humans and the limits of outlier approaches
Abstract
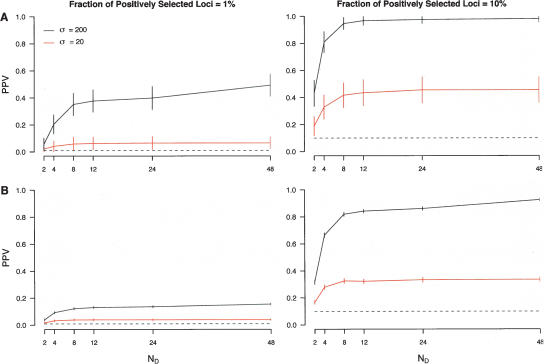
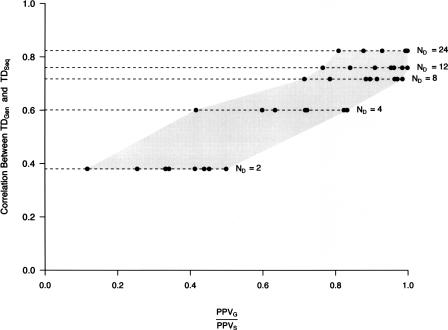
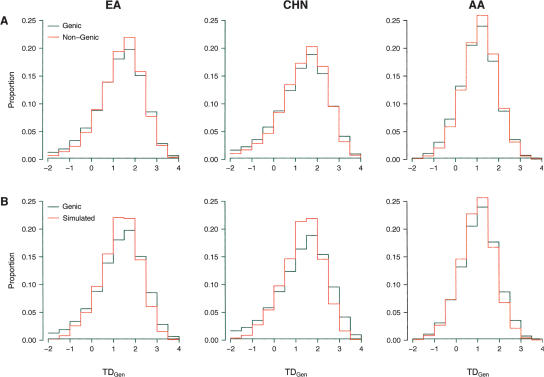
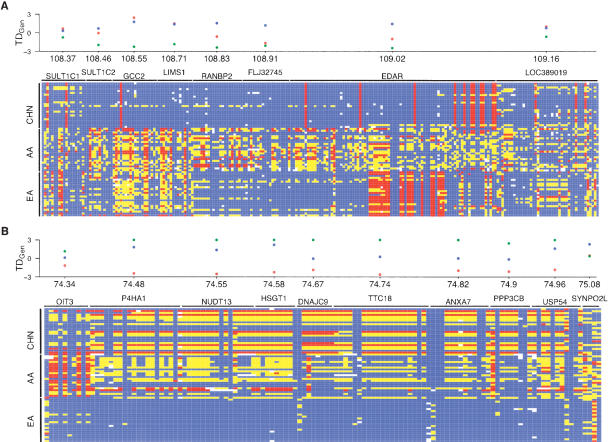
Identifying regions of the human genome that have been targets of positive selection will provide important insights into recent human evolutionary history and may facilitate the search for complex disease genes. However, the confounding effects of population demographic history and selection on patterns of genetic variation complicate inferences of selection when a small number of loci are studied. To this end, identifying outlier loci from empirical genome-wide distributions of genetic variation is a promising strategy to detect targets of selection. Here, we evaluate the power and efficiency of a simple outlier approach and describe a genome-wide scan for positive selection using a dense catalog of 1.58 million SNPs that were genotyped in three human populations. In total, we analyzed 14,589 genes, 385 of which possess patterns of genetic variation consistent with the hypothesis of positive selection. Furthermore, several extended genomic regions were found, spanning >500 kb, that contained multiple contiguous candidate selection genes. More generally, these data provide important practical insights into the limits of outlier approaches in genome-wide scans for selection, provide strong candidate selection genes to study in greater detail, and may have important implications for disease related research.
Figures
References
-
- Akey J.M., Zhang K., Xiong M., Jin L., Zhang K., Xiong M., Jin L., Xiong M., Jin L., Jin L. The effect of single nucleotide polymorphism identification strategies on estimates of linkage disequilibrium. Mol. Biol. Evol. 2003;20:232–242. - PubMed
-
- Akey J.M., Eberle M.A., Rieder M.J., Carlson C.S., Shriver M.D., Nickerson D.A., Kruglyak L., Eberle M.A., Rieder M.J., Carlson C.S., Shriver M.D., Nickerson D.A., Kruglyak L., Rieder M.J., Carlson C.S., Shriver M.D., Nickerson D.A., Kruglyak L., Carlson C.S., Shriver M.D., Nickerson D.A., Kruglyak L., Shriver M.D., Nickerson D.A., Kruglyak L., Nickerson D.A., Kruglyak L., Kruglyak L. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004;2:e286. - PMC - PubMed
-
- Andolfatto P. Adaptive hitchhiking effects on genome variability. Curr. Opin. Genet. Dev. 2001;11:635–641. - PubMed
-
- Bamshad M., Wooding S.P., Wooding S.P. Signatures of natural selection in the human genome. Nat. Rev. Genet. 2003;4:99–111. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources