

The Alexander disease-causing glial fibrillary acidic protein mutant, R416W, accumulates into Rosenthal fibers by a pathway that involves filament aggregation and the association of alpha B-crystallin and HSP27
- PMID: 16826512
- PMCID: PMC1559481
- DOI: 10.1086/504411
The Alexander disease-causing glial fibrillary acidic protein mutant, R416W, accumulates into Rosenthal fibers by a pathway that involves filament aggregation and the association of alpha B-crystallin and HSP27
Abstract
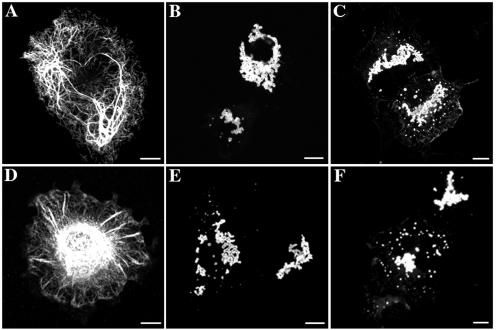
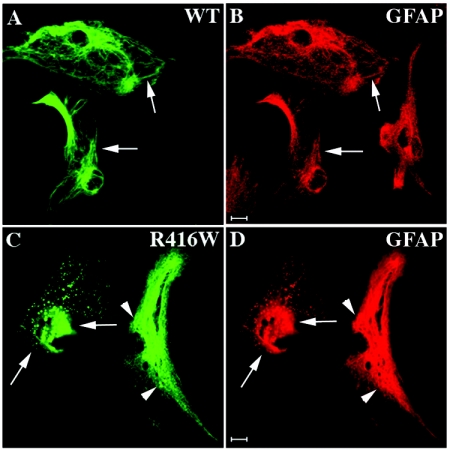
Here, we describe the early events in the disease pathogenesis of Alexander disease. This is a rare and usually fatal neurodegenerative disorder whose pathological hallmark is the abundance of protein aggregates in astrocytes. These aggregates, termed "Rosenthal fibers," contain the protein chaperones alpha B-crystallin and HSP27 as well as glial fibrillary acidic protein (GFAP), an intermediate filament (IF) protein found almost exclusively in astrocytes. Heterozygous, missense GFAP mutations that usually arise spontaneously during spermatogenesis have recently been found in the majority of patients with Alexander disease. In this study, we show that one of the more frequently observed mutations, R416W, significantly perturbs in vitro filament assembly. The filamentous structures formed resemble assembly intermediates but aggregate more strongly. Consistent with the heterozygosity of the mutation, this effect is dominant over wild-type GFAP in coassembly experiments. Transient transfection studies demonstrate that R416W GFAP induces the formation of GFAP-containing cytoplasmic aggregates in a wide range of different cell types, including astrocytes. The aggregates have several important features in common with Rosenthal fibers, including the association of alpha B-crystallin and HSP27. This association occurs simultaneously with the formation of protein aggregates containing R416W GFAP and is also specific, since HSP70 does not partition with them. Monoclonal antibodies specific for R416W GFAP reveal, for the first time for any IF-based disease, the presence of the mutant protein in the characteristic histopathological feature of the disease, namely Rosenthal fibers. Collectively, these data confirm that the effects of the R416W GFAP are dominant, changing the assembly process in a way that encourages aberrant filament-filament interactions that then lead to protein aggregation and chaperone sequestration as early events in Alexander disease.
Figures








Similar articles
-
Alexander disease causing mutations in the C-terminal domain of GFAP are deleterious both to assembly and network formation with the potential to both activate caspase 3 and decrease cell viability.Exp Cell Res. 2011 Oct 1;317(16):2252-66. doi: 10.1016/j.yexcr.2011.06.017. Epub 2011 Jul 2. Exp Cell Res. 2011. PMID: 21756903 Free PMC article.
-
Synemin is expressed in reactive astrocytes and Rosenthal fibers in Alexander disease.APMIS. 2014 Jan;122(1):76-80. doi: 10.1111/apm.12088. Epub 2013 Apr 18. APMIS. 2014. PMID: 23594359
-
Beneficial effects of curcumin on GFAP filament organization and down-regulation of GFAP expression in an in vitro model of Alexander disease.Exp Cell Res. 2012 Sep 10;318(15):1844-54. doi: 10.1016/j.yexcr.2012.06.008. Epub 2012 Jun 15. Exp Cell Res. 2012. PMID: 22705585
-
GFAP mutations in Alexander disease.Int J Dev Neurosci. 2002 Jun-Aug;20(3-5):259-68. doi: 10.1016/s0736-5748(02)00019-9. Int J Dev Neurosci. 2002. PMID: 12175861 Review.
-
GFAP and its role in Alexander disease.Exp Cell Res. 2007 Jun 10;313(10):2077-87. doi: 10.1016/j.yexcr.2007.04.004. Epub 2007 Apr 6. Exp Cell Res. 2007. PMID: 17498694 Free PMC article. Review.
Cited by
-
Genotypic and phenotypic heterogeneity among Chinese pediatric genetic white matter disorders.Ital J Pediatr. 2023 Nov 19;49(1):155. doi: 10.1186/s13052-023-01555-z. Ital J Pediatr. 2023. PMID: 37981684 Free PMC article.
-
Identification of a novel nonsense mutation in the rod domain of GFAP that is associated with Alexander disease.Eur J Hum Genet. 2015 Jan;23(1):72-8. doi: 10.1038/ejhg.2014.68. Epub 2014 Apr 23. Eur J Hum Genet. 2015. PMID: 24755947 Free PMC article.
-
"Toxic memory" via chaperone modification is a potential mechanism for rapid Mallory-Denk body reinduction.Hepatology. 2008 Sep;48(3):931-42. doi: 10.1002/hep.22430. Hepatology. 2008. PMID: 18697205 Free PMC article.
-
Glial fibrillary acidic protein filaments can tolerate the incorporation of assembly-compromised GFAP-delta, but with consequences for filament organization and alphaB-crystallin association.Mol Biol Cell. 2008 Oct;19(10):4521-33. doi: 10.1091/mbc.e08-03-0284. Epub 2008 Aug 6. Mol Biol Cell. 2008. PMID: 18685083 Free PMC article.
-
Human iPSC-Derived Astrocytes: A Powerful Tool to Study Primary Astrocyte Dysfunction in the Pathogenesis of Rare Leukodystrophies.Int J Mol Sci. 2021 Dec 27;23(1):274. doi: 10.3390/ijms23010274. Int J Mol Sci. 2021. PMID: 35008700 Free PMC article. Review.
References
Web Resources
-
- Alexander Disease Web site, http://www.waisman.wisc.edu/alexander/home.htmlx
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for GFAP [accession number J04569])
-
- Intermediate Filament Disease Mutation Database, http://www.interfil.org/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Alexander disease)
References
-
- Alexander WS (1949) Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 72:373–381 - PubMed
-
- Russo LS Jr, Aron A, Anderson PJ (1976) Alexander’s disease: a report and reappraisal. Neurology 26:607–614 - PubMed
-
- Neal JW, Cave EM, Singhrao SK, Cole G, Wallace SJ (1992) Alexander’s disease in infancy and childhood: a report of two cases. Acta Neuropathol (Berl) 84:322–327 - PubMed
-
- Deprez M, D’Hooghe M, Misson JP, de Leval L, Ceuterick C, Reznik M, Martin JJ, D’Hooge M (1999) Infantile and juvenile presentations of Alexander’s disease: a report of two cases. Acta Neurol Scand 99:158–165 - PubMed
-
- Rodriguez D, Gauthier F, Bertini E, Bugiani M, Brenner M, N’Guyen S, Goizet C, Gelot A, Surtees R, Pedespan J-M, Hernandorena X, Troncoso M, Uziel G, Messing A, Ponsot G, Pham-Dinh D, Dautigny A, Boespflug-Tanguy O (2001) Infantile Alexander disease: spectrum of GFAP mutations and genotype-phenotype correlation. Am J Hum Genet 69:1134–1140 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
