

Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites
- PMID: 16826513
- PMCID: PMC1559486
- DOI: 10.1086/505407
Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites
Abstract
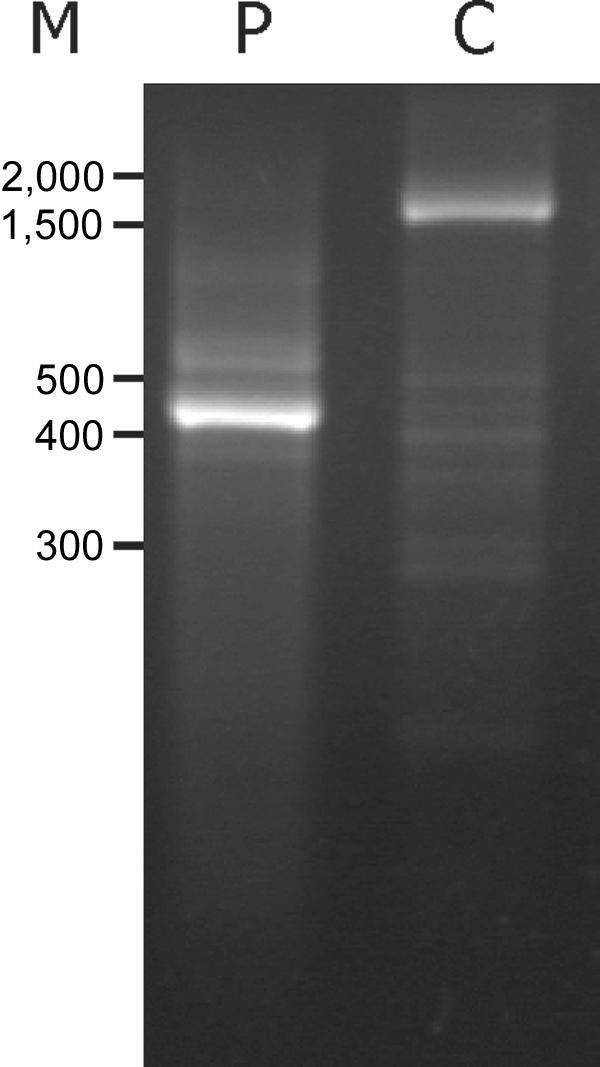
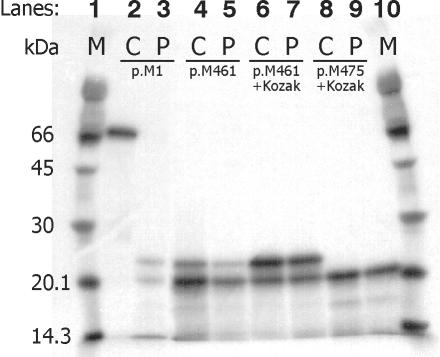
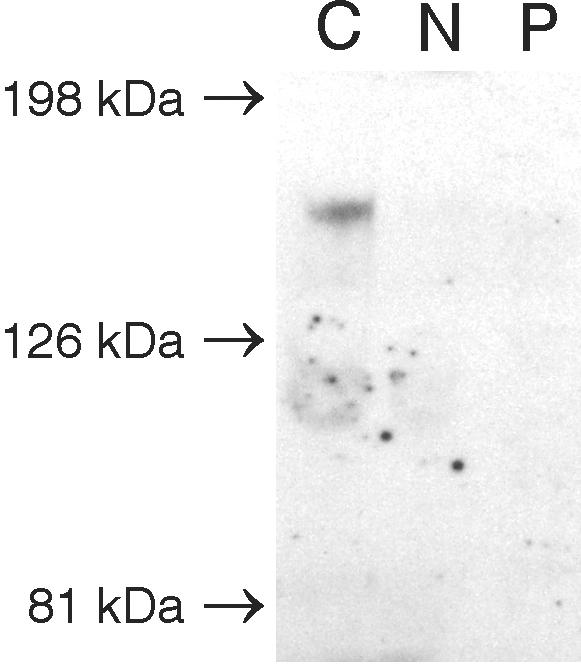
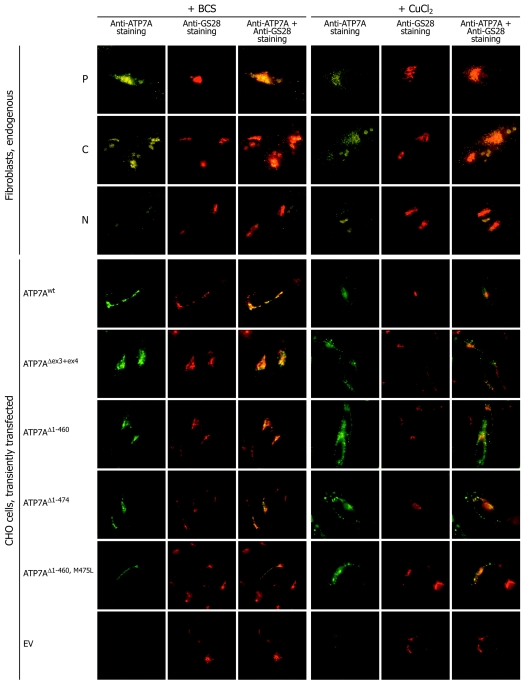
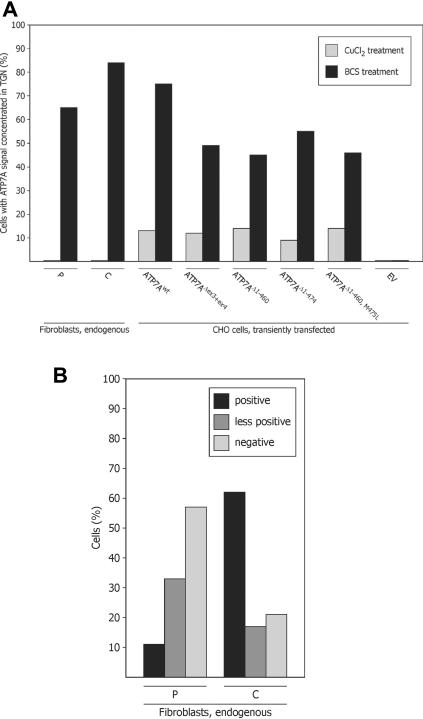
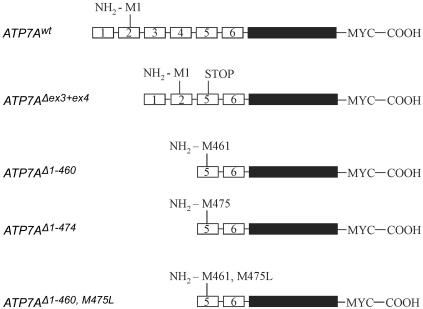
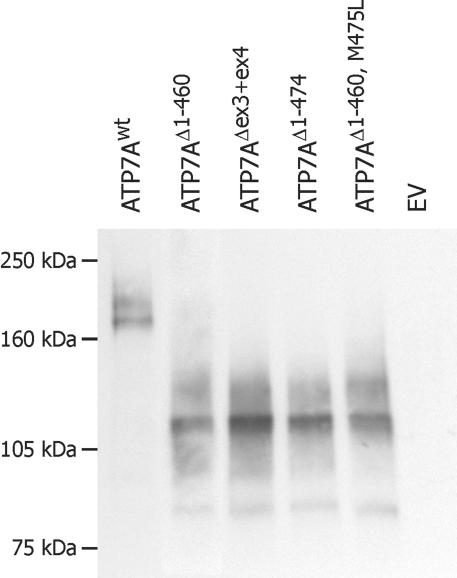
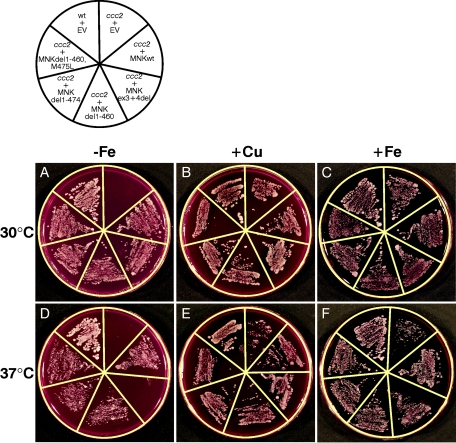
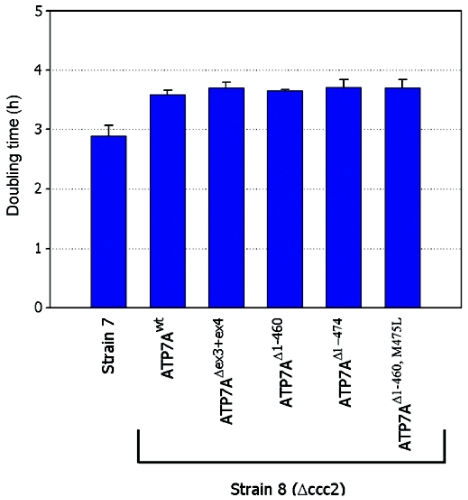
Menkes disease (MD) is an X-linked recessive disorder of copper metabolism. It is caused by mutations in the ATP7A gene encoding a copper-translocating P-type ATPase, which contains six N-terminal copper-binding sites (CBS1-CBS6). Most patients die in early childhood. We investigated the functional effect of a large frameshift deletion in ATP7A (including exons 3 and 4) identified in a patient with MD with unexpectedly mild symptoms and long survival. The mutated transcript, ATP7A(Delta ex3+ex4), contains a premature termination codon after 46 codons. Although such transcripts are generally degraded by nonsense-mediated mRNA decay (NMD), it was established by real-time PCR quantification that the ATP7A(Delta ex3+ex4) transcript was protected from degradation. A combination of in vitro translation, recombinant expression, and immunocytochemical analysis provided evidence that the ATP7A(Delta ex3+ex4) transcript was protected from degradation because of reinitiation of protein translation. Our findings suggest that reinitiation takes place at two downstream internal codons. The putative N-terminally truncated proteins contain only CBS5 and CBS6. Cellular localization and copper-dependent trafficking of the major part of endogenous and recombinant ATP7A(Delta ex3+ex4) proteins were similar to the wild-type ATP7A protein. Furthermore, the ATP7A(Delta ex3+ex4) cDNA was able to rescue a yeast strain lacking the homologous gene, CCC2. In summary, we propose that reinitiation of the NMD-resistant ATP7A(Delta ex3+ex4) transcript leads to the synthesis of N-terminally truncated and at-least-partially functional Menkes proteins missing CBS1-CBS4. This finding--that a mutation that would have been assumed to be null is not--highlights the need to examine the biochemical phenotype of patients to deduce the efficacy of copper therapy.
Figures

Similar articles
-
A novel two-nucleotide deletion in the ATP7A gene associated with delayed infantile onset of Menkes disease.Pediatr Neurol. 2014 Apr;50(4):417-20. doi: 10.1016/j.pediatrneurol.2014.01.005. Epub 2014 Jan 5. Pediatr Neurol. 2014. PMID: 24630286 Free PMC article.
-
A novel frameshift mutation in exon 23 of ATP7A (MNK) results in occipital horn syndrome and not in Menkes disease.Am J Hum Genet. 2001 Aug;69(2):420-7. doi: 10.1086/321290. Epub 2001 Jun 26. Am J Hum Genet. 2001. PMID: 11431706 Free PMC article.
-
Small amounts of functional ATP7A protein permit mild phenotype.J Trace Elem Med Biol. 2015;31:173-7. doi: 10.1016/j.jtemb.2014.07.022. Epub 2014 Aug 8. J Trace Elem Med Biol. 2015. PMID: 25172213 Review.
-
Mutation in the CPC motif-containing 6th transmembrane domain affects intracellular localization, trafficking and copper transport efficiency of ATP7A protein in mosaic mutant mice--an animal model of Menkes disease.Metallomics. 2012 Feb;4(2):197-204. doi: 10.1039/c1mt00134e. Epub 2011 Nov 16. Metallomics. 2012. PMID: 22089129
-
Menkes copper-translocating P-type ATPase (ATP7A): biochemical and cell biology properties, and role in Menkes disease.J Bioenerg Biomembr. 2002 Oct;34(5):363-71. doi: 10.1023/a:1021250003104. J Bioenerg Biomembr. 2002. PMID: 12539963 Review.
Cited by
-
How to get away with nonsense: Mechanisms and consequences of escape from nonsense-mediated RNA decay.Wiley Interdiscip Rev RNA. 2020 Jan;11(1):e1560. doi: 10.1002/wrna.1560. Epub 2019 Jul 29. Wiley Interdiscip Rev RNA. 2020. PMID: 31359616 Free PMC article. Review.
-
LQT2 nonsense mutations generate trafficking defective NH2-terminally truncated channels by the reinitiation of translation.Am J Physiol Heart Circ Physiol. 2013 Nov 1;305(9):H1397-404. doi: 10.1152/ajpheart.00304.2013. Epub 2013 Aug 30. Am J Physiol Heart Circ Physiol. 2013. PMID: 23997099 Free PMC article.
-
13 novel putative mutations in ATP7A found in a cohort of 25 Italian families.Metab Brain Dis. 2017 Aug;32(4):1173-1183. doi: 10.1007/s11011-017-0010-8. Epub 2017 Apr 28. Metab Brain Dis. 2017. PMID: 28451781
-
Ohtahara syndrome in a family with an ARX protein truncation mutation (c.81C>G/p.Y27X).Eur J Hum Genet. 2010 Feb;18(2):157-62. doi: 10.1038/ejhg.2009.139. Epub 2009 Sep 9. Eur J Hum Genet. 2010. PMID: 19738637 Free PMC article.
-
Translation reinitiation after uORFs does not fully protect mRNAs from nonsense-mediated decay.RNA. 2023 Jun;29(6):735-744. doi: 10.1261/rna.079525.122. Epub 2023 Mar 6. RNA. 2023. PMID: 36878710 Free PMC article.
References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for ATP7A [accession number L06133.1] and ATP7A [accession number NP_000043])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MD, ATP7A, WD, and ATP7B)
References
-
- Møller JV, Juul B, Le Maire M (1996) Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim Biophys Acta 1286:1–51 - PubMed
-
- Horn N, Tümer Z (2002) Menkes disease and the occipital horn syndrome. In: Royce P, Steinmann B (eds) Connective tissue and its heritable disorders, 2nd ed. Wiley-Liss, New York, pp 651–685
-
- Cox DW (1996) Molecular advances in Wilson disease. Prog Liver Dis 14:245–264 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
