

A coalescence-guided hierarchical Bayesian method for haplotype inference
- PMID: 16826521
- PMCID: PMC1559491
- DOI: 10.1086/506276
A coalescence-guided hierarchical Bayesian method for haplotype inference
Abstract
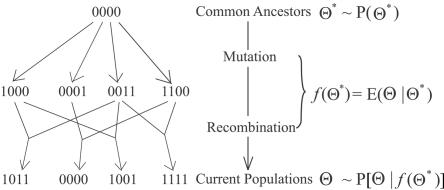
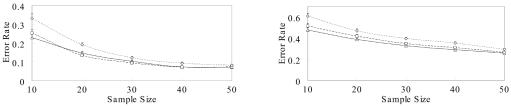
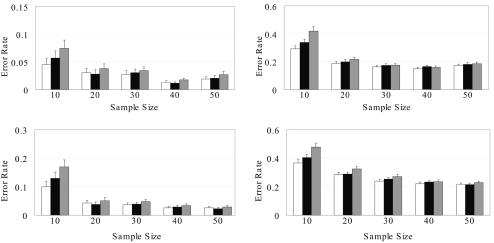
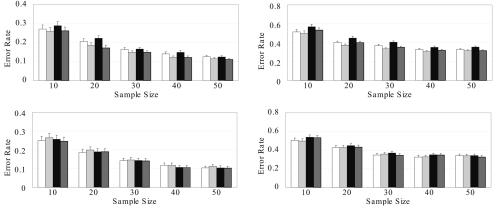
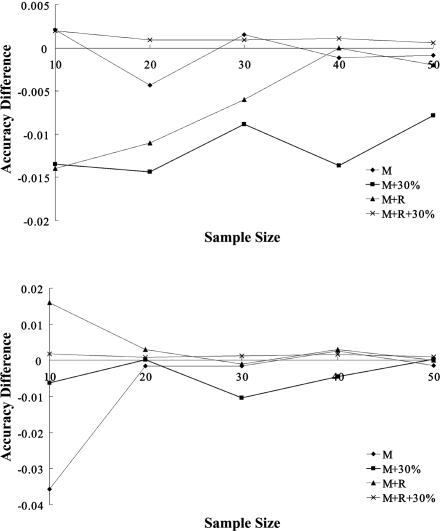
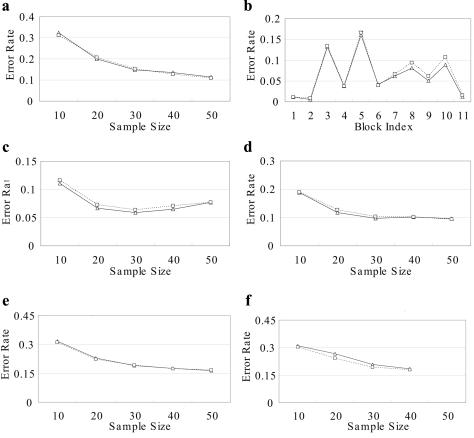
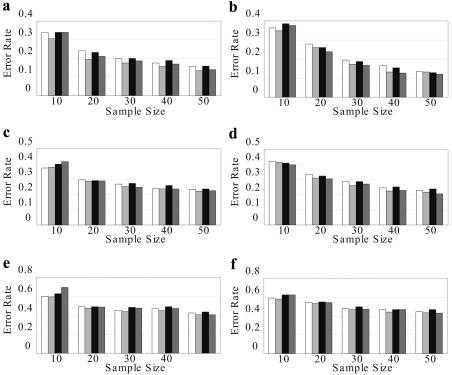
Haplotype inference from phase-ambiguous multilocus genotype data is an important task for both disease-gene mapping and studies of human evolution. We report a novel haplotype-inference method based on a coalescence-guided hierarchical Bayes model. In this model, a hierarchical structure is imposed on the prior haplotype frequency distributions to capture the similarities among modern-day haplotypes attributable to their common ancestry. As a consequence, the model both allows distinct haplotypes to have different a priori probabilities according to the inferred hierarchical ancestral structure and results in a proper joint posterior distribution for all the parameters of interest. A Markov chain-Monte Carlo scheme is designed to draw from this posterior distribution. By using coalescence-based simulation and empirically generated data sets (Whitehead Institute's inflammatory bowel disease data sets and HapMap data sets), we demonstrate the merits of the new method in comparison with HAPLOTYPER and PHASE, with or without the presence of recombination hotspots and missing genotypes.
Figures





References
Web Resources
-
- Coalescence-guided Hierarchical Bayesian Model for Haplotype Inference, http://www.people.fas.harvard.edu/~junliu/chb/ (for supplementary materials, detailed documentation, and download instructions for CHB algorithm)
-
- International HapMap Project, http://www.hapmap.org/
-
- ms: A program for generating samples under neutral models, http://home.uchicago.edu/~rhudson1/source/mksamples.html (for Hudson's program)
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
