

Emergence of protein fold families through rational design
- PMID: 16839198
- PMCID: PMC1487181
- DOI: 10.1371/journal.pcbi.0020085
Emergence of protein fold families through rational design
Abstract
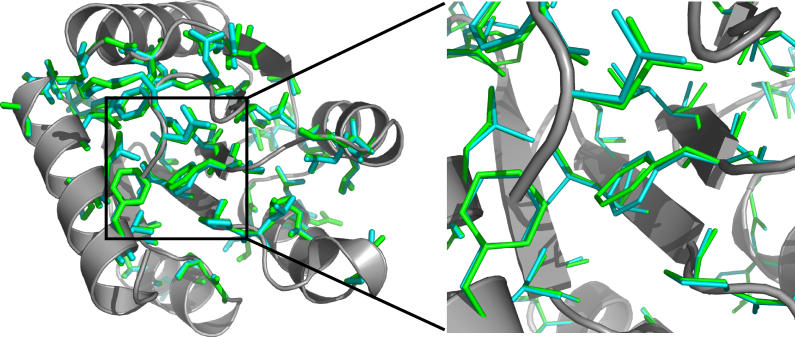
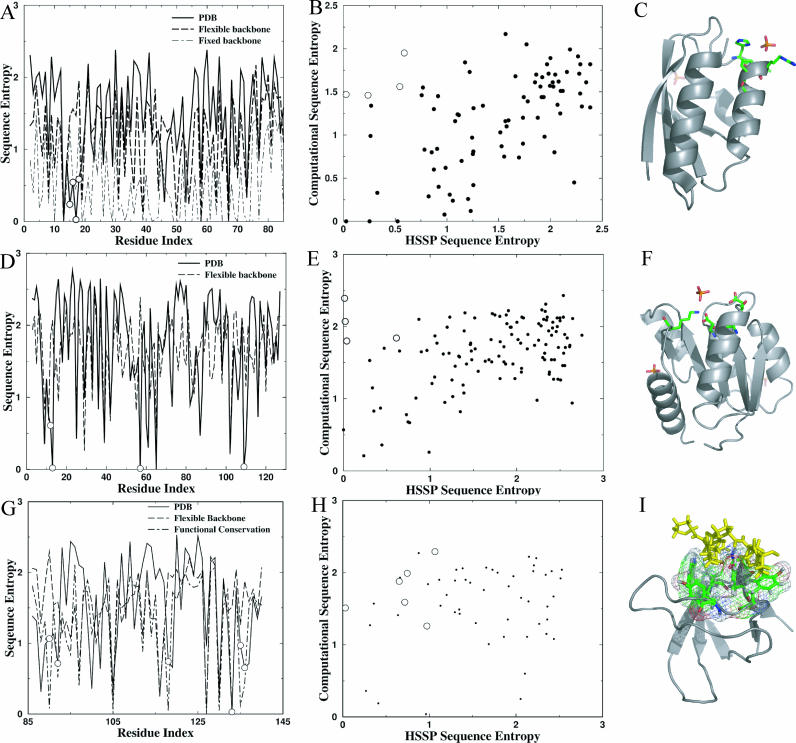
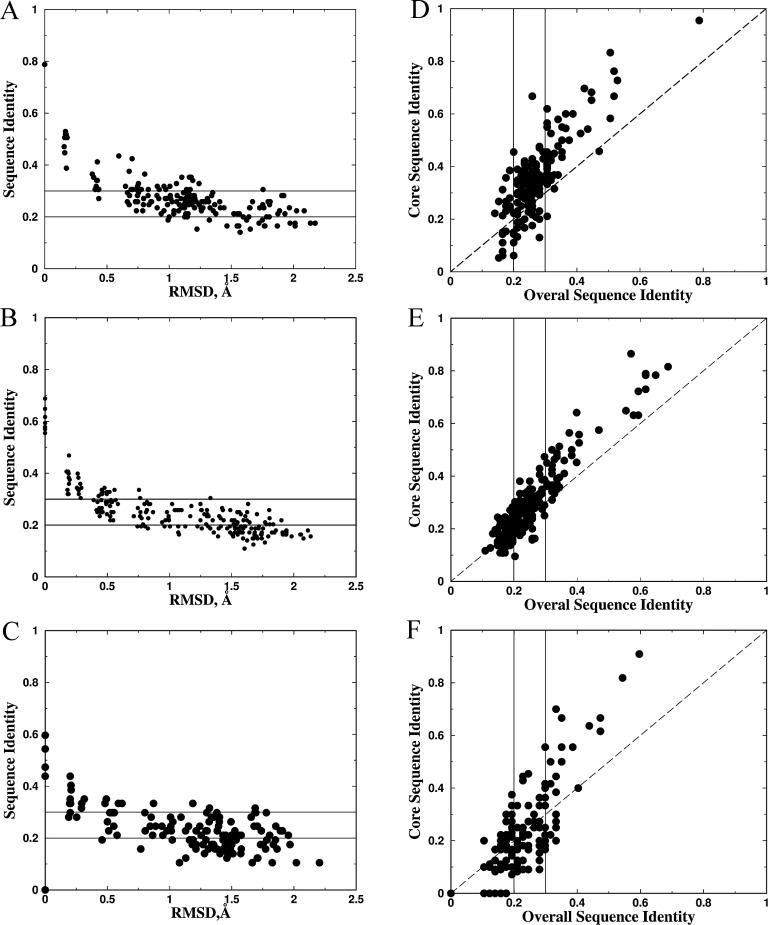
Diverse proteins with similar structures are grouped into families of homologs and analogs, if their sequence similarity is higher or lower, respectively, than 20%-30%. It was suggested that protein homologs and analogs originate from a common ancestor and diverge in their distinct evolutionary time scales, emerging as a consequence of the physical properties of the protein sequence space. Although a number of studies have determined key signatures of protein family organization, the sequence-structure factors that differentiate the two evolution-related protein families remain unknown. Here, we stipulate that subtle structural changes, which appear due to accumulating mutations in the homologous families, lead to distinct packing of the protein core and, thus, novel compositions of core residues. The latter process leads to the formation of distinct families of homologs. We propose that such differentiation results in the formation of analogous families. To test our postulate, we developed a molecular modeling and design toolkit, Medusa, to computationally design protein sequences that correspond to the same fold family. We find that analogous proteins emerge when a backbone structure deviates only 1-2 angstroms root-mean-square deviation from the original structure. For close homologs, core residues are highly conserved. However, when the overall sequence similarity drops to approximately 25%-30%, the composition of core residues starts to diverge, thereby forming novel families of protein homologs. This direct observation of the formation of protein homologs within a specific fold family supports our hypothesis. The conservation of amino acids in designed sequences recapitulates that of the naturally occurring sequences, thereby validating our computational design methodology.
Conflict of interest statement
Figures
Similar articles
-
The ConSurf-HSSP database: the mapping of evolutionary conservation among homologs onto PDB structures.Proteins. 2005 Feb 15;58(3):610-7. doi: 10.1002/prot.20305. Proteins. 2005. PMID: 15614759
-
Realm of PD-(D/E)XK nuclease superfamily revisited: detection of novel families with modified transitive meta profile searches.BMC Struct Biol. 2007 Jun 20;7:40. doi: 10.1186/1472-6807-7-40. BMC Struct Biol. 2007. PMID: 17584917 Free PMC article.
-
Identification of homologous core structures.Proteins. 1999 Apr 1;35(1):70-9. Proteins. 1999. PMID: 10090287
-
Fold change in evolution of protein structures.J Struct Biol. 2001 May-Jun;134(2-3):167-85. doi: 10.1006/jsbi.2001.4335. J Struct Biol. 2001. PMID: 11551177 Review.
-
From protein structure to function.Curr Opin Struct Biol. 1999 Jun;9(3):374-82. doi: 10.1016/S0959-440X(99)80051-7. Curr Opin Struct Biol. 1999. PMID: 10361094 Review.
Cited by
-
The structural heterogeneity of α-synuclein is governed by several distinct subpopulations with interconversion times slower than milliseconds.Structure. 2021 Sep 2;29(9):1048-1064.e6. doi: 10.1016/j.str.2021.05.002. Epub 2021 May 19. Structure. 2021. PMID: 34015255 Free PMC article.
-
β-Methylamino-L-alanine substitution of serine in SOD1 suggests a direct role in ALS etiology.PLoS Comput Biol. 2019 Jul 19;15(7):e1007225. doi: 10.1371/journal.pcbi.1007225. eCollection 2019 Jul. PLoS Comput Biol. 2019. PMID: 31323035 Free PMC article.
-
Toward the Accuracy and Speed of Protein Side-Chain Packing: A Systematic Study on Rotamer Libraries.J Chem Inf Model. 2020 Jan 27;60(1):410-420. doi: 10.1021/acs.jcim.9b00812. Epub 2019 Dec 31. J Chem Inf Model. 2020. PMID: 31851497 Free PMC article.
-
Atomic interaction networks in the core of protein domains and their native folds.PLoS One. 2010 Feb 23;5(2):e9391. doi: 10.1371/journal.pone.0009391. PLoS One. 2010. PMID: 20186337 Free PMC article.
-
A generic program for multistate protein design.PLoS One. 2011;6(7):e20937. doi: 10.1371/journal.pone.0020937. Epub 2011 Jul 6. PLoS One. 2011. PMID: 21754981 Free PMC article.
References
-
- Levitt M, Chothia C. Structural patterns in globular proteins. Nature. 1976;261:552–558. - PubMed
-
- Orengo CA, Jones DT, Thornton JM. Protein superfamilies and domain superfolds. Nature. 1994;372:631–634. - PubMed
-
- Govindarajan S, Goldstein RA. The foldability landscape of model proteins. Biopolymers. 1997;42:427–438. - PubMed
-
- Finkelstein AV, Gutun AM, Badretdinov AY. Why are the same protein folds used to perform different functions? FEBS Lett. 1993;325:23–28. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
