

Prevalence and genetic diversity of coronaviruses in bats from China
- PMID: 16840328
- PMCID: PMC1563713
- DOI: 10.1128/JVI.00697-06
Prevalence and genetic diversity of coronaviruses in bats from China
Abstract
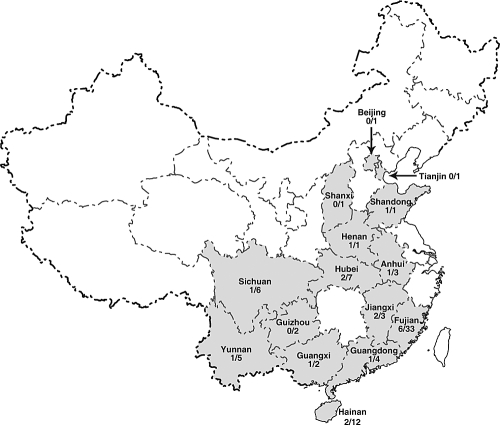
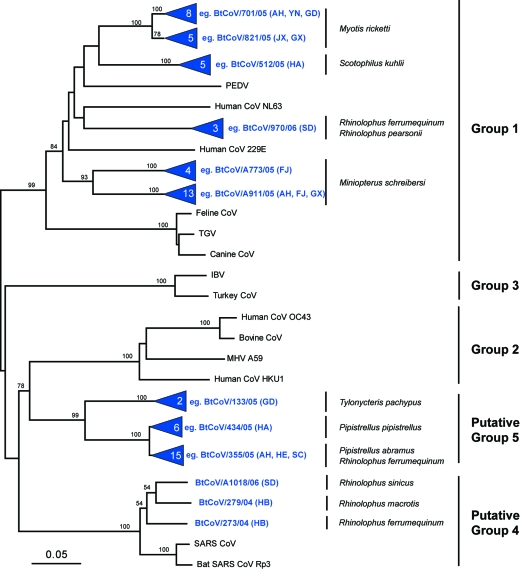
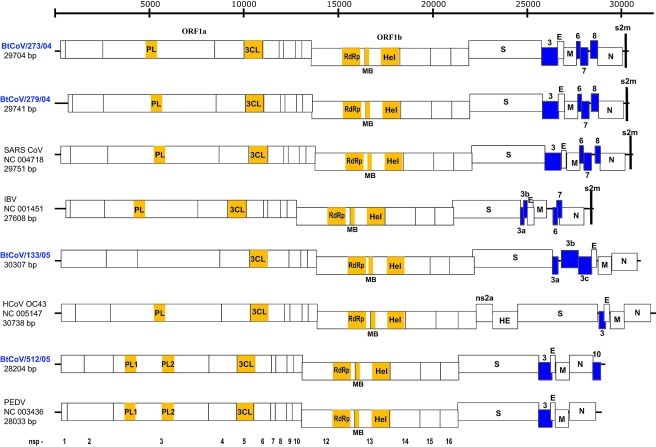
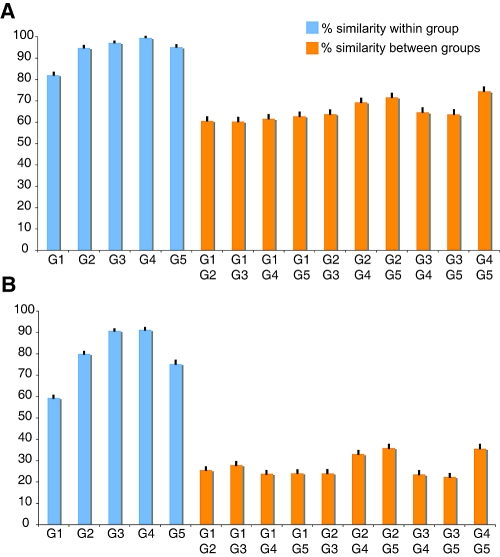
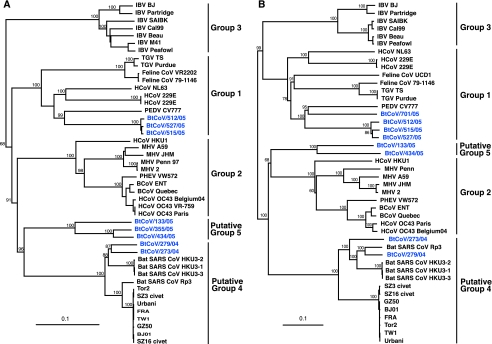
Coronaviruses can infect a variety of animals including poultry, livestock, and humans and are currently classified into three groups. The interspecies transmissions of coronaviruses between different hosts form a complex ecosystem of which little is known. The outbreak of severe acute respiratory syndrome (SARS) and the recent identification of new coronaviruses have highlighted the necessity for further investigation of coronavirus ecology, in particular the role of bats and other wild animals. In this study, we sampled bat populations in 15 provinces of China and reveal that approximately 6.5% of the bats, from diverse species distributed throughout the region, harbor coronaviruses. Full genomes of four coronavirues from bats were sequenced and analyzed. Phylogenetic analyses of the spike, envelope, membrane, and nucleoprotein structural proteins and the two conserved replicase domains, putative RNA-dependent RNA polymerase and RNA helicase, revealed that bat coronaviruses cluster in three different groups: group 1, another group that includes all SARS and SARS-like coronaviruses (putative group 4), and an independent bat coronavirus group (putative group 5). Further genetic analyses showed that different species of bats maintain coronaviruses from different groups and that a single bat species from different geographic locations supports similar coronaviruses. Thus, the findings of this study suggest that bats may play an integral role in the ecology and evolution of coronaviruses.
Figures
References
-
- Altringham, J. D. 1996. Bats: biology and behavior, p. 1-48. Oxford University Press, Oxford, United Kingdom.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
