

Multiplex sequencing of paired-end ditags (MS-PET): a strategy for the ultra-high-throughput analysis of transcriptomes and genomes
- PMID: 16840528
- PMCID: PMC1524903
- DOI: 10.1093/nar/gkl444
Multiplex sequencing of paired-end ditags (MS-PET): a strategy for the ultra-high-throughput analysis of transcriptomes and genomes
Abstract
The paired-end ditagging (PET) technique has been shown to be efficient and accurate for large-scale transcriptome and genome analysis. However, as with other DNA tag-based sequencing strategies, it is constrained by the current efficiency of Sanger technology. A recently developed multiplex sequencing method (454-sequencing) using picolitre-scale reactions has achieved a remarkable advance in efficiency, but suffers from short-read lengths, and a lack of paired-end information. To further enhance the efficiency of PET analysis and at the same time overcome the drawbacks of the new sequencing method, we coupled multiplex sequencing with paired-end ditagging (MS-PET) using modified PET procedures to simultaneously sequence 200,000 to 300,000 dimerized PET (diPET) templates, with an output of nearly half-a-million PET sequences in a single 4 h machine run. We demonstrate the utility and robustness of MS-PET by analyzing the transcriptome of human breast carcinoma cells, and by mapping p53 binding sites in the genome of human colorectal carcinoma cells. This combined sequencing strategy achieved an approximate 100-fold efficiency increase over the current standard for PET analysis, and furthermore enables the short-read-length multiplex sequencing procedure to acquire paired-end information from large DNA fragments.
Figures
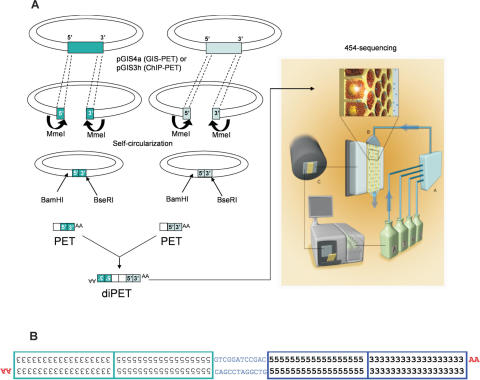
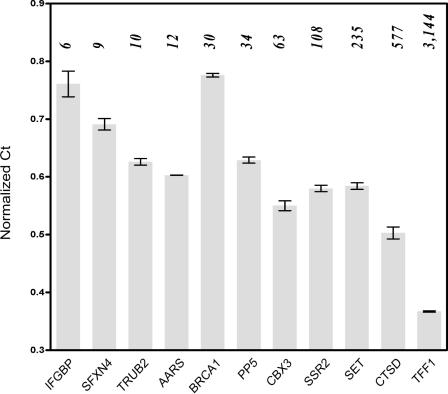
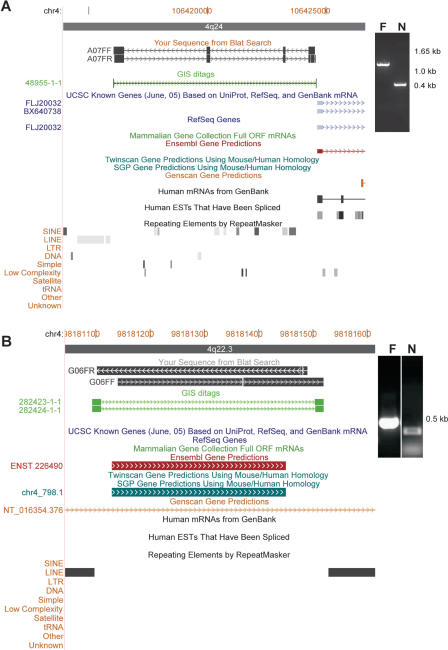
References
-
- The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. - PubMed
-
- Velculescu V.E., Zhang L., Vogelstein B., Kinzler K.W. Serial analysis of gene expression. Science. 1995;270:484–487. - PubMed
-
- Saha S., Sparks A.B., Rago C., Akmaev V., Wang C.J., Vogelstein B., Kinzler K.W., Velculescu V.E. Using the transcriptome to annotate the genome. Nat. Biotechnol. 2002;20:508–512. - PubMed
-
- Shiraki T., Kondo S., Katayama S., Waki K., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T., et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl Acad. Sci. USA. 2003;100:15776–15781. - PMC - PubMed
-
- Hashimoto S., Suzuki Y., Kasai Y., Morohoshi K., Yamada T., Sese J., Morishita S., Sugano S., Matsushima K. 5′ End SAGE for the analysis of transcriptional start sites. Nat. Biotechnol. 2004;22:1146–1149. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
