

Mapping of protein disulfide bonds using negative ion fragmentation with a broadband precursor selection
- PMID: 16841900
- PMCID: PMC2505194
- DOI: 10.1021/ac060132w
Mapping of protein disulfide bonds using negative ion fragmentation with a broadband precursor selection
Abstract
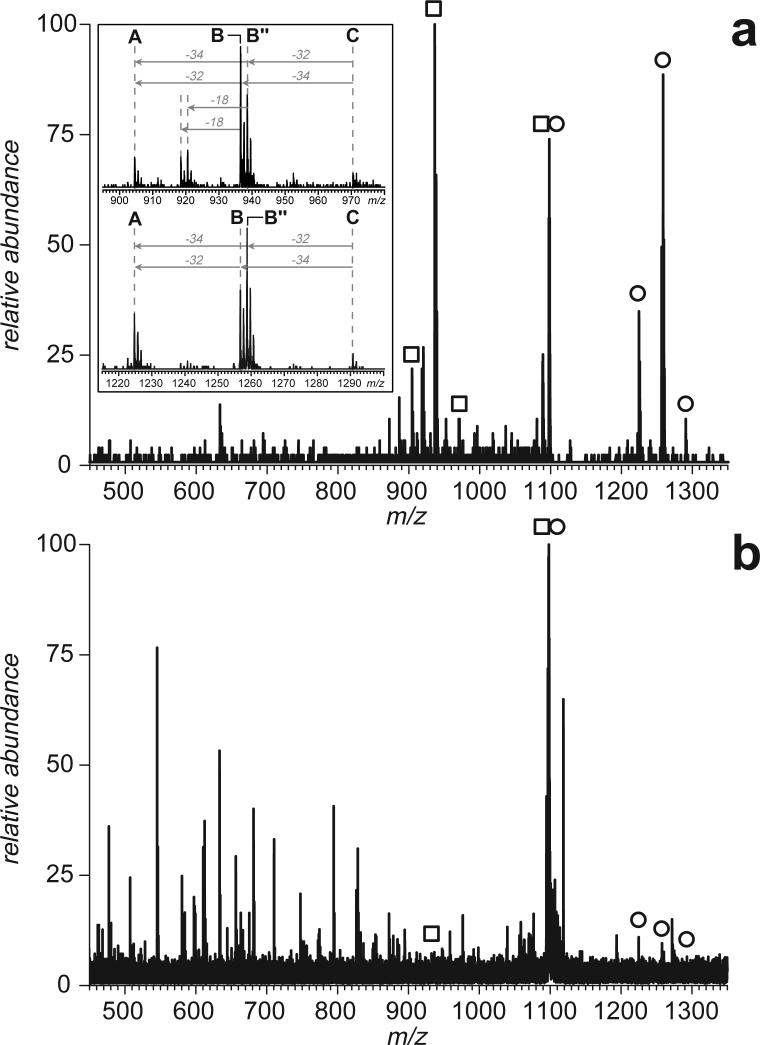
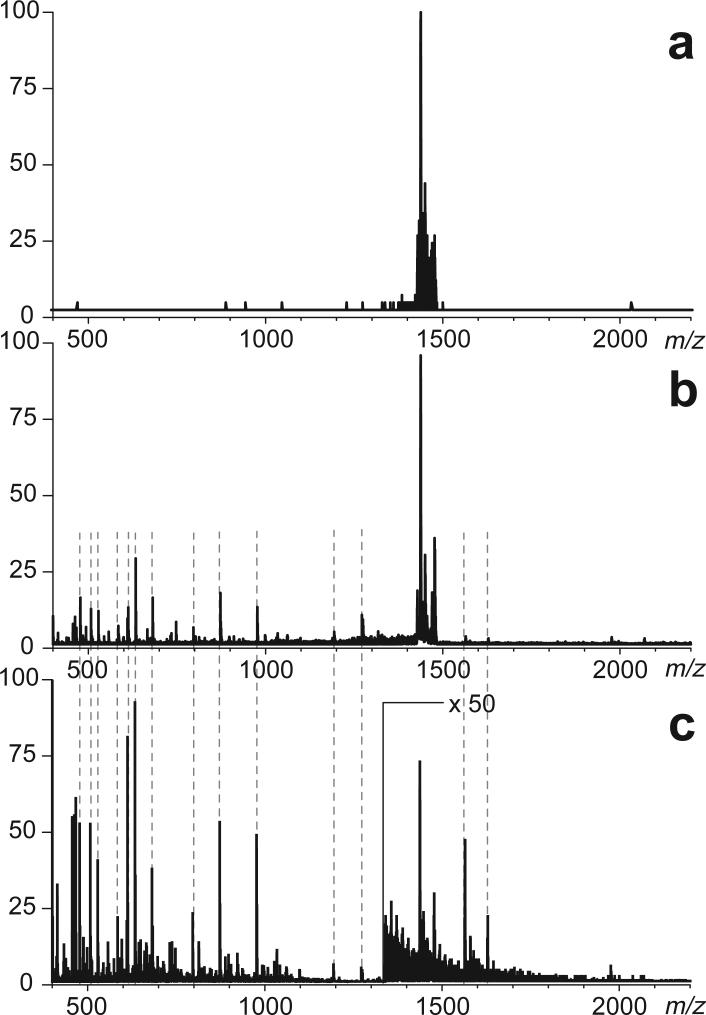
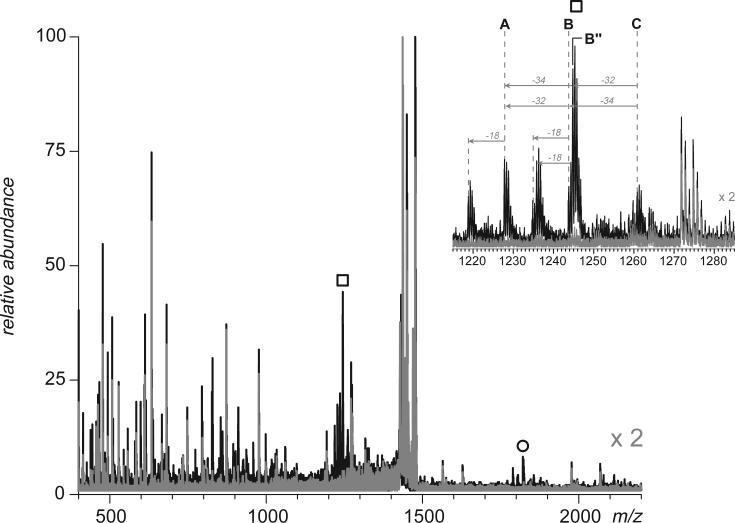
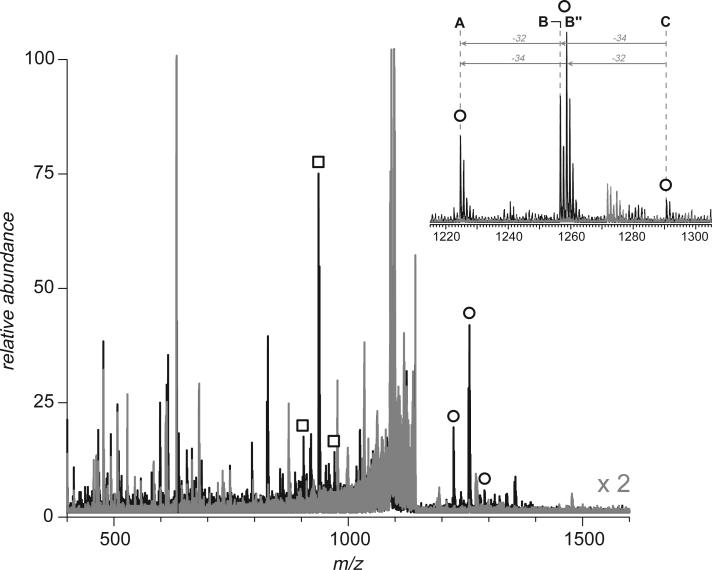
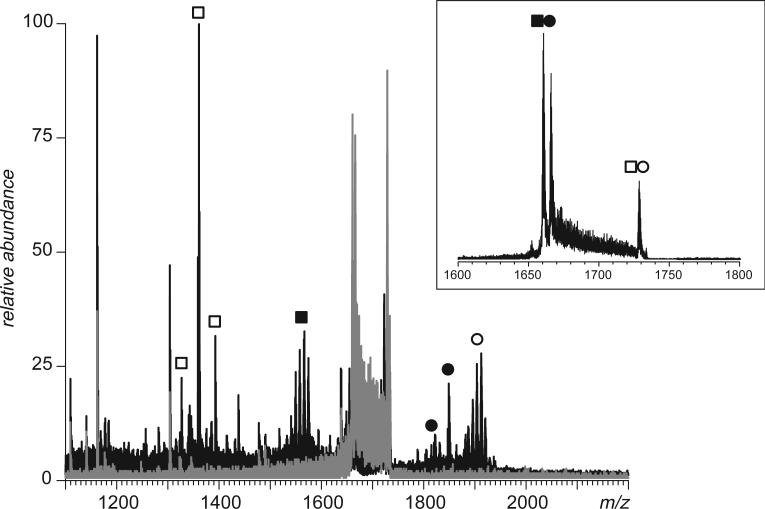
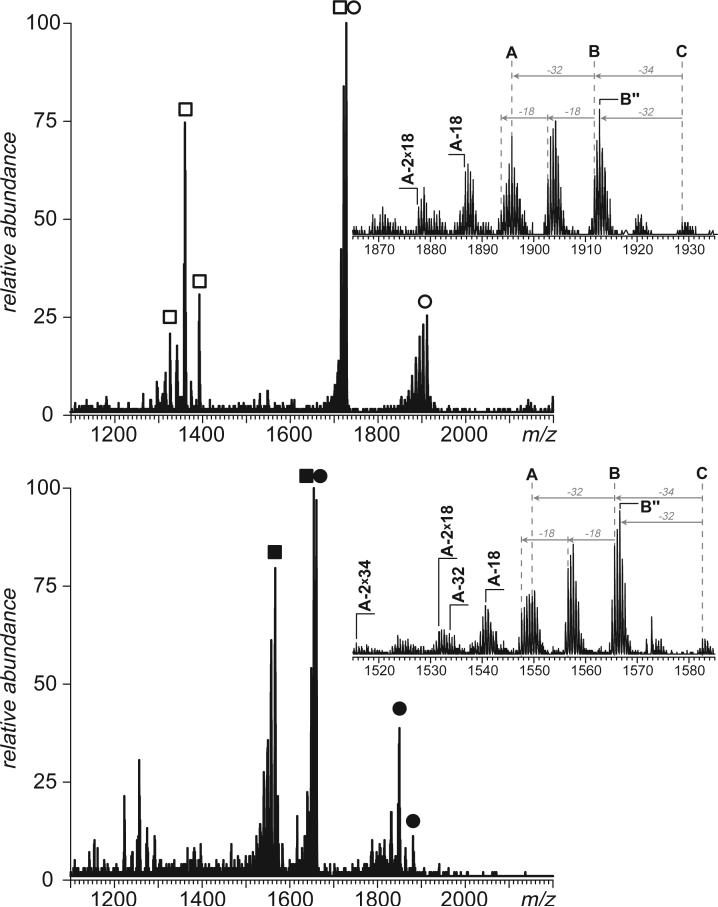
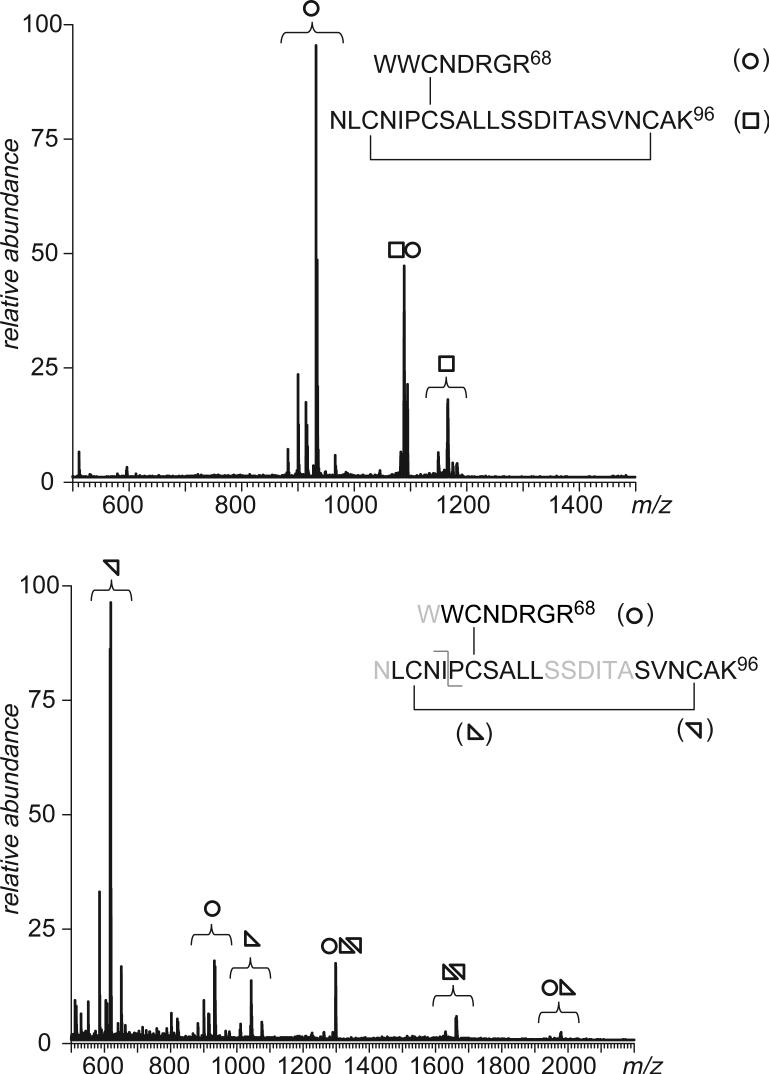
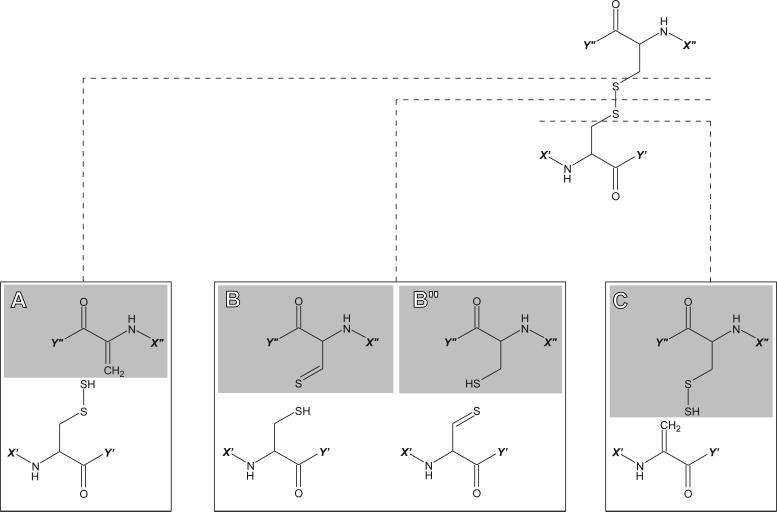
Fast mapping of disulfide bonds in proteins containing multiple cysteine residues is often required in order to assess the integrity of the tertiary structure of proteins prone to degradation and misfolding or to detect distinct intermediate states generated in the course of oxidative folding. A new method of rapid detection and identification of disulfide-linked peptides in complex proteolytic mixtures utilizes the tendency of collision-activated peptide ions to lose preferentially side chains of select amino acids in the negative ion mode. Cleavages of cysteine side chains result in efficient dissociation of disulfide bonds and produce characteristic signatures in the fragment ion mass spectra. While cleavages of other side chains result in insignificant loss of mass and full retention of the peptide ion charge, dissociation of external disulfide bonds results in physical separation of two peptides and, therefore, significant changes of both mass and charge of fragment ions relative to the precursor ion. This feature allows the fragment ions generated by dissociation of external disulfide bonds to be easily detected and identified even if multiple precursor ions are activated simultaneously. Such broadband selection of precursor ions for consecutive activation is achieved by lowering the dc/rf amplitude ratio in the first quadrupole filter of a hybrid quadrupole time-of-flight mass spectrometer. The feasibility of the new method is demonstrated by partial mapping of disulfide bridges within a 37-kDa protein containing 16 cysteine residues and complete disulfide mapping within a lysozyme (14.5 kDa) containing 8 cysteine residues. In addition to detecting peptide pairs connected by a single external disulfide, the new method is also shown to be capable of identifying peptides containing both external and internal disulfide bonds. The two major factors determining the efficiency of disulfide mapping using the new methodology are the effectiveness of proteolysis and the ability of the resulting proteolytic fragments to form multiply charged negative ions.
Figures
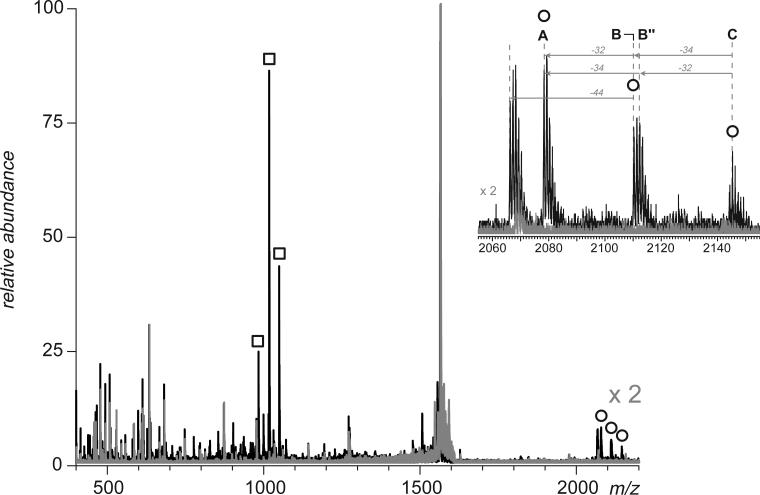
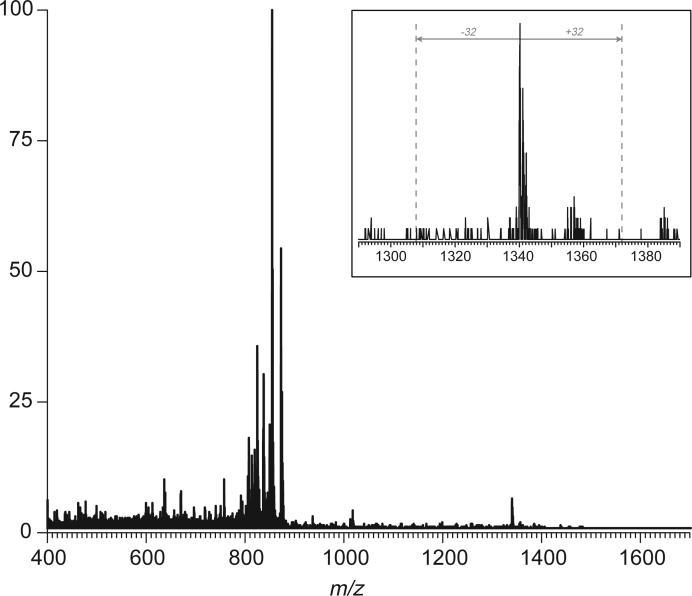
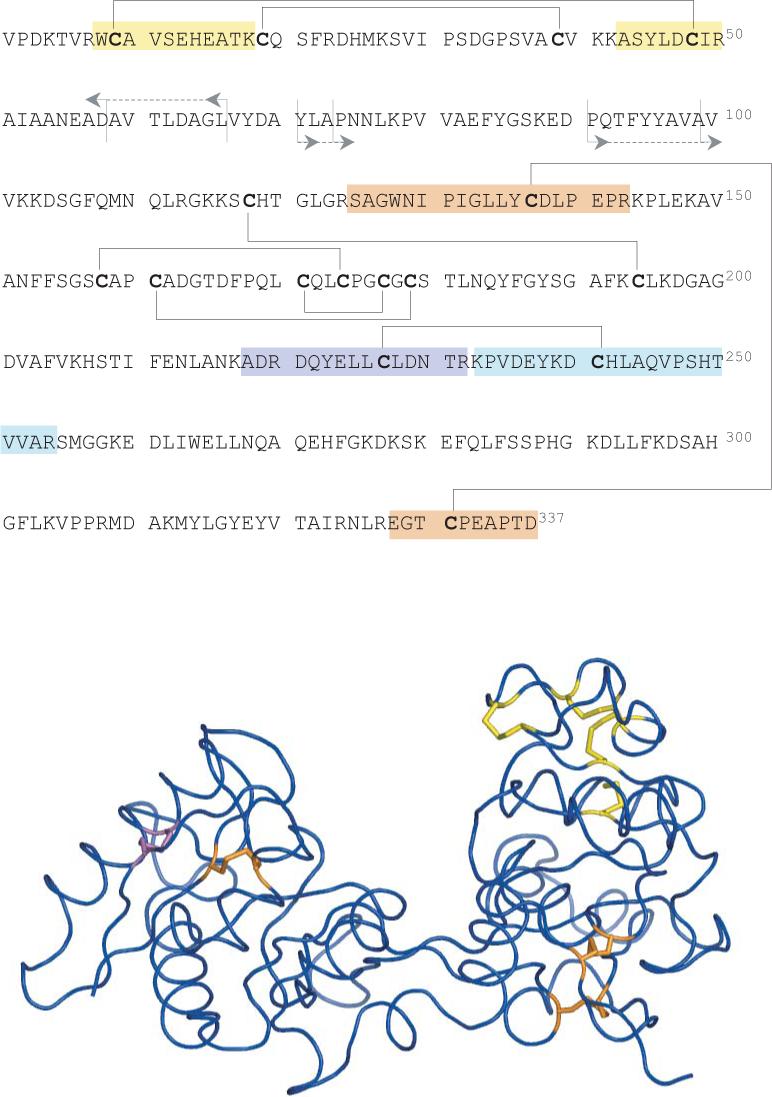
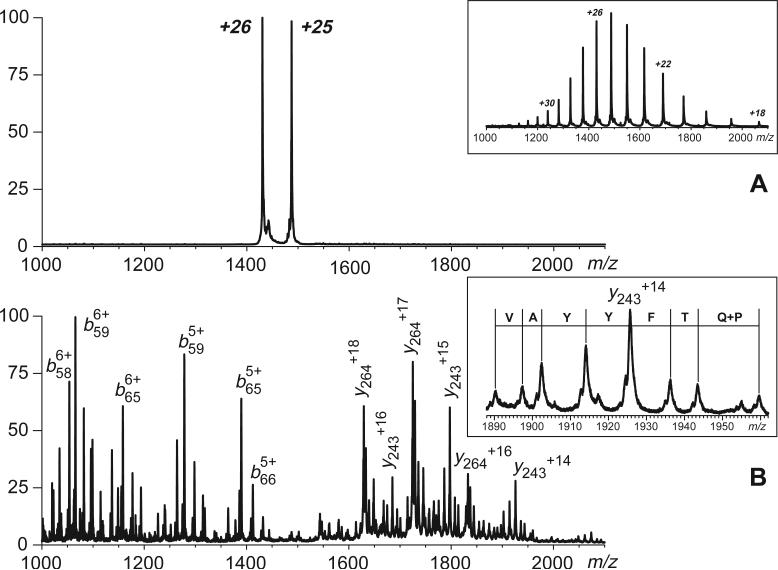
References
-
- Standing KG. Peptide and protein de novo sequencing by mass spectrometry. Curr. Opin. Struct. Biol. 2003;13:595–601. - PubMed
-
- Coon JJ, Syka JE, Shabanowitz J, Hunt DF. Tandem mass spectrometry for peptide and protein sequence analysis. Biotechniques. 2005;38:519–523. - PubMed
-
- Kaltashov IA, Eyles SJ. Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spectrom. Rev. 2002;21:37–71. - PubMed
-
- Welker E, Wedemeyer WJ, Narayan M, Scheraga HA. Coupling of conformational folding and disulfide-bond reactions in oxidative folding of proteins. Biochemistry. 2001;40:9059–9064. - PubMed
-
- Yu J. Intentionally degrading protein pharmaceuticals to validate stability-indicating analytical methods. BioPharm. Appl. 2000;13:46–50.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
