

GeneAlign: a coding exon prediction tool based on phylogenetical comparisons
- PMID: 16845010
- PMCID: PMC1538901
- DOI: 10.1093/nar/gkl307
GeneAlign: a coding exon prediction tool based on phylogenetical comparisons
Abstract
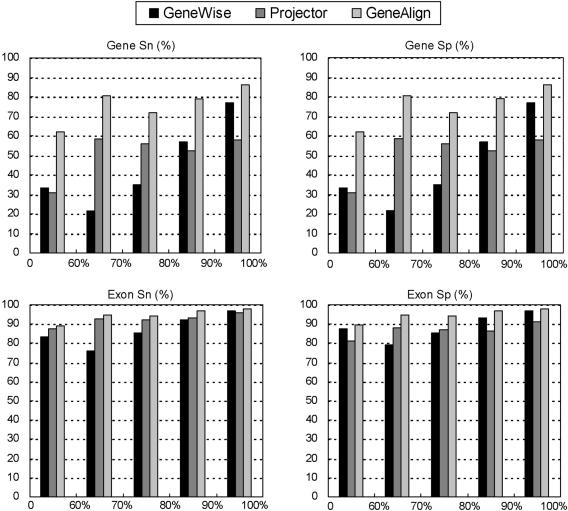
GeneAlign is a coding exon prediction tool for predicting protein coding genes by measuring the homologies between a sequence of a genome and related sequences, which have been annotated, of other genomes. Identifying protein coding genes is one of most important tasks in newly sequenced genomes. With increasing numbers of gene annotations verified by experiments, it is feasible to identify genes in the newly sequenced genomes by comparing to annotated genes of phylogenetically close organisms. GeneAlign applies CORAL, a heuristic linear time alignment tool, to determine if regions flanked by the candidate signals (initiation codon-GT, AG-GT and AG-STOP codon) are similar to annotated coding exons. Employing the conservation of gene structures and sequence homologies between protein coding regions increases the prediction accuracy. GeneAlign was tested on Projector dataset of 491 human-mouse homologous sequence pairs. At the gene level, both the average sensitivity and the average specificity of GeneAlign are 81%, and they are larger than 96% at the exon level. The rates of missing exons and wrong exons are smaller than 1%. GeneAlign is a free tool available at http://genealign.hccvs.hc.edu.tw.
Figures
References
-
- Brent M.R., Buigo R. Recent advances in gene structure prediction. Curr. Opin. Struct. Biol. 2004;14:264–272. - PubMed
-
- Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. - PubMed
-
- Brendel V., Xing L., Zhu W. Gene structure prediction from consensus spliced alignment of multiple ESTs matching the same genomic locus. Bioinformatics. 2004;20:1157–1169. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
