

NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes
- PMID: 16845035
- PMCID: PMC1538769
- DOI: 10.1093/nar/gkl244
NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes
Abstract
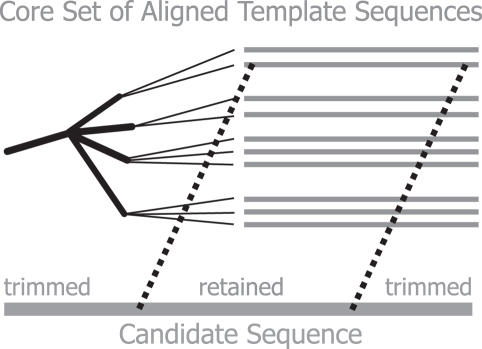
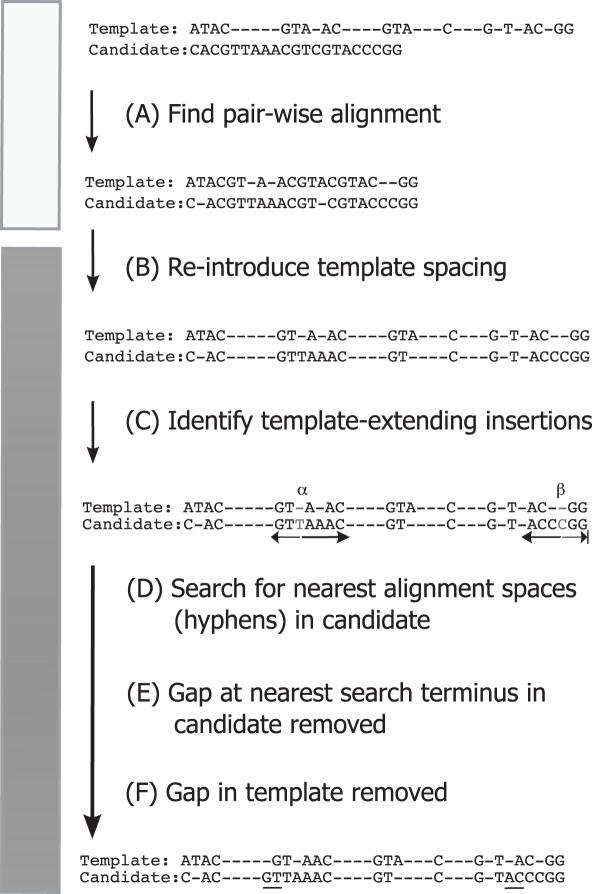
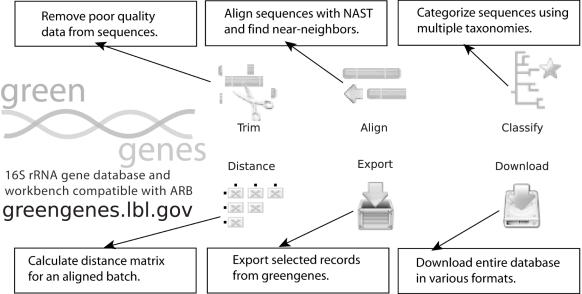
Microbiologists conducting surveys of bacterial and archaeal diversity often require comparative alignments of thousands of 16S rRNA genes collected from a sample. The computational resources and bioinformatics expertise required to construct such an alignment has inhibited high-throughput analysis. It was hypothesized that an online tool could be developed to efficiently align thousands of 16S rRNA genes via the NAST (Nearest Alignment Space Termination) algorithm for creating multiple sequence alignments (MSA). The tool was implemented with a web-interface at http://greengenes.lbl.gov/NAST. Each user-submitted sequence is compared with Greengenes' 'Core Set', comprising approximately 10,000 aligned non-chimeric sequences representative of the currently recognized diversity among bacteria and archaea. User sequences are oriented and paired with their closest match in the Core Set to serve as a template for inserting gap characters. Non-16S data (sequence from vector or surrounding genomic regions) are conveniently removed in the returned alignment. From the resulting MSA, distance matrices can be calculated for diversity estimates and organisms can be classified by taxonomy. The ability to align and categorize large sequence sets using a simple interface has enabled researchers with various experience levels to obtain bacterial and archaeal community profiles.
Figures
References
-
- Fox G.E., Stackebrandt E., Hespell R.B., Gibson J., Maniloff J., Dyer T.A., Wolfe R.S., Balch W.E., Tanner R.S., Magrum L.J., et al. The phylogeny of prokaryotes. Science. 1980;209:457–463. - PubMed
-
- Woese C.R., Fox G.E., Zablen L., Uchida T., Bonen L., Pechman K., Lewis B.J., Stahl D. Conservation of primary structure in 16S ribosomal RNA. Nature. 1975;254:83–86. - PubMed
-
- Kong Y., Ong S.L., Ng W.J., Liu W.T. Diversity and distribution of a deeply branched novel proteobacterial group found in anaerobic–aerobic activated sludge processes. Environ. Microbiol. 2002;4:753–757. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
