

NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis
- PMID: 16845062
- PMCID: PMC1538881
- DOI: 10.1093/nar/gkl082
NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis
Abstract
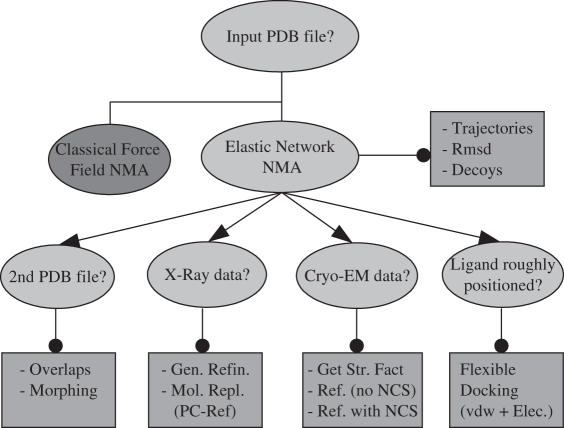
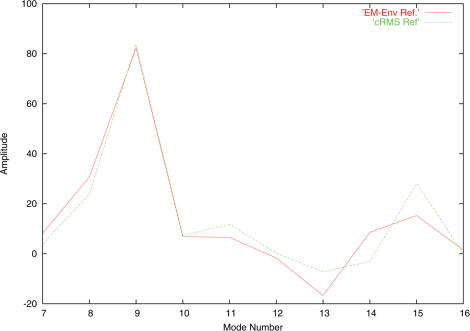
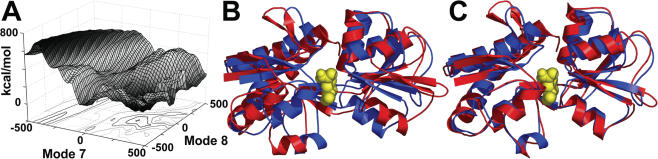
Normal mode analysis (NMA) is an efficient way to study collective motions in biomolecules that bypasses the computational costs and many limitations associated with full dynamics simulations. The NOMAD-Ref web server presented here provides tools for online calculation of the normal modes of large molecules (up to 100,000 atoms) maintaining a full all-atom representation of their structures, as well as access to a number of programs that utilize these collective motions for deformation and refinement of biomolecular structures. Applications include the generation of sets of decoys with correct stereochemistry but arbitrary large amplitude movements, the quantification of the overlap between alternative conformations of a molecule, refinement of structures against experimental data, such as X-ray diffraction structure factors or Cryo-EM maps and optimization of docked complexes by modeling receptor/ligand flexibility through normal mode motions. The server can be accessed at the URL http://lorentz.immstr.pasteur.fr/nomad-ref.php.
Figures
Similar articles
-
Refinement of docked protein-ligand and protein-DNA structures using low frequency normal mode amplitude optimization.Nucleic Acids Res. 2005 Aug 8;33(14):4496-506. doi: 10.1093/nar/gki730. Print 2005. Nucleic Acids Res. 2005. PMID: 16087736 Free PMC article.
-
ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement.Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W610-4. doi: 10.1093/nar/gkh368. Nucleic Acids Res. 2004. PMID: 15215461 Free PMC article.
-
WEBnm@: a web application for normal mode analyses of proteins.BMC Bioinformatics. 2005 Mar 11;6:52. doi: 10.1186/1471-2105-6-52. BMC Bioinformatics. 2005. PMID: 15762993 Free PMC article.
-
Computational Methodologies for Real-Space Structural Refinement of Large Macromolecular Complexes.Annu Rev Biophys. 2016 Jul 5;45:253-78. doi: 10.1146/annurev-biophys-062215-011113. Epub 2016 May 2. Annu Rev Biophys. 2016. PMID: 27145875 Free PMC article. Review.
-
Strategies for crystallization and structure determination of very large macromolecular assemblies.Curr Opin Struct Biol. 2007 Oct;17(5):572-9. doi: 10.1016/j.sbi.2007.09.004. Epub 2007 Oct 25. Curr Opin Struct Biol. 2007. PMID: 17964135 Review.
Cited by
-
PredyFlexy: flexibility and local structure prediction from sequence.Nucleic Acids Res. 2012 Jul;40(Web Server issue):W317-22. doi: 10.1093/nar/gks482. Epub 2012 Jun 11. Nucleic Acids Res. 2012. PMID: 22689641 Free PMC article.
-
A novel computational and structural analysis of nsSNPs in CFTR gene.Genomic Med. 2008 Jan;2(1-2):23-32. doi: 10.1007/s11568-008-9019-8. Epub 2008 May 14. Genomic Med. 2008. PMID: 18716917 Free PMC article.
-
Association of functional variants and protein-to-protein physical interactions of human MutY homolog linked with familial adenomatous polyposis and colorectal cancer syndrome.Noncoding RNA Res. 2019 Dec 4;4(4):155-173. doi: 10.1016/j.ncrna.2019.11.005. eCollection 2019 Dec. Noncoding RNA Res. 2019. PMID: 32072083 Free PMC article.
-
X-ray structure of the pestivirus NS3 helicase and its conformation in solution.J Virol. 2015 Apr;89(8):4356-71. doi: 10.1128/JVI.03165-14. Epub 2015 Feb 4. J Virol. 2015. PMID: 25653438 Free PMC article.
-
In silico profiling of nonsynonymous SNPs of fat mass and obesity-associated gene: possible impacts on the treatment of non-alcoholic fatty liver disease.Lipids Health Dis. 2023 Jan 30;22(1):17. doi: 10.1186/s12944-023-01782-7. Lipids Health Dis. 2023. PMID: 36717943 Free PMC article.
References
-
- Wodak S.J., Mendez R. Predictions of protein–protein interactions: the CAPRI experiment, its evaluation and implications. Curr. Opin. Struct. Biol. 2004;14:242–249. - PubMed
-
- Tirion M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996;77:1905–1908. - PubMed
-
- Bahar I., Atligan A.R., Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2:173–181. - PubMed
-
- Hinsen K. Analysis of domain motions by approximate normal mode calculations. Proteins: Struct. Funct. and Genet. 1998;33:417–429. - PubMed
-
- Krebs W.G., Alexandrov V., Wilson C.A., Echols N., Yu H., Gerstein M. Normal mode analysis of macromolecular motions in a database framework: developing mode concentration as a useful classifying statistic. Proteins: Struct. Funct. and Genet. 2002;48:682–695. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
