

Selective inactivation of glutaredoxin by sporidesmin and other epidithiopiperazinediones
- PMID: 16846241
- PMCID: PMC3199604
- DOI: 10.1021/bi060440o
Selective inactivation of glutaredoxin by sporidesmin and other epidithiopiperazinediones
Abstract
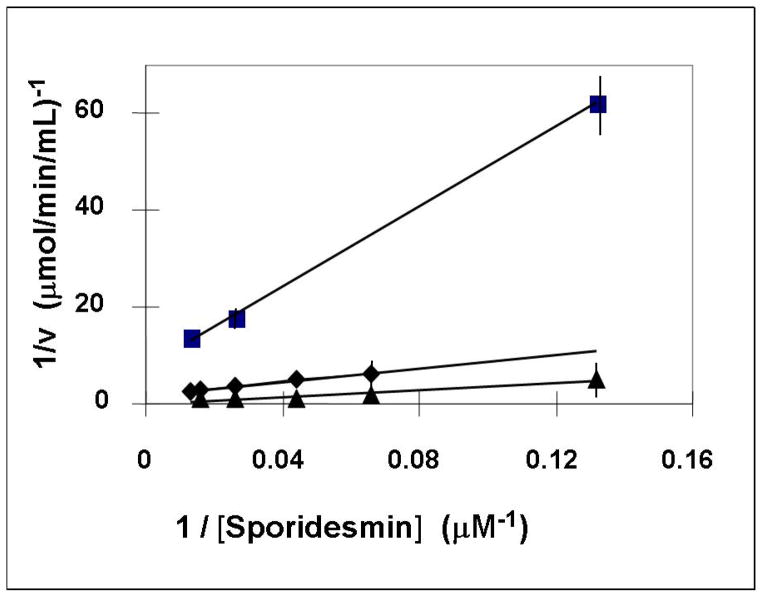
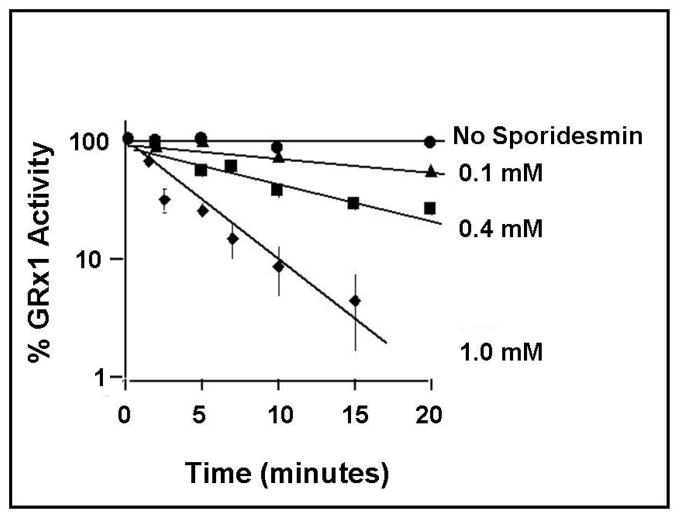
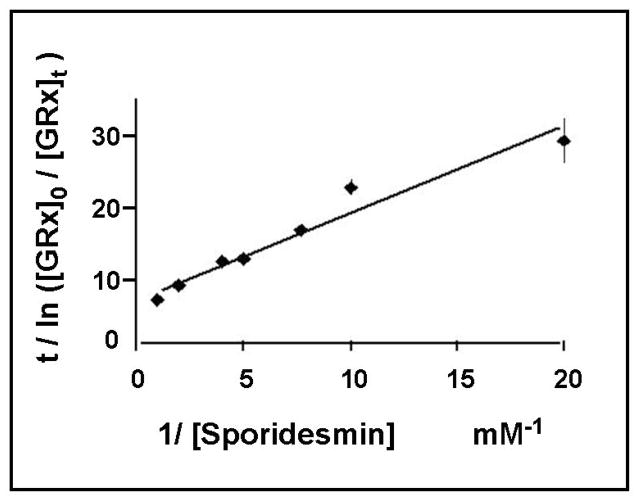
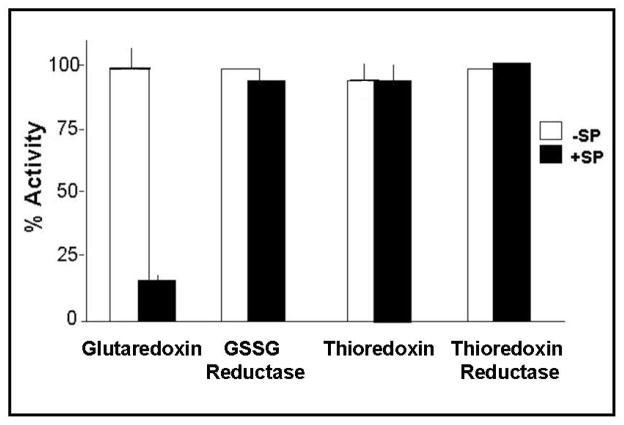
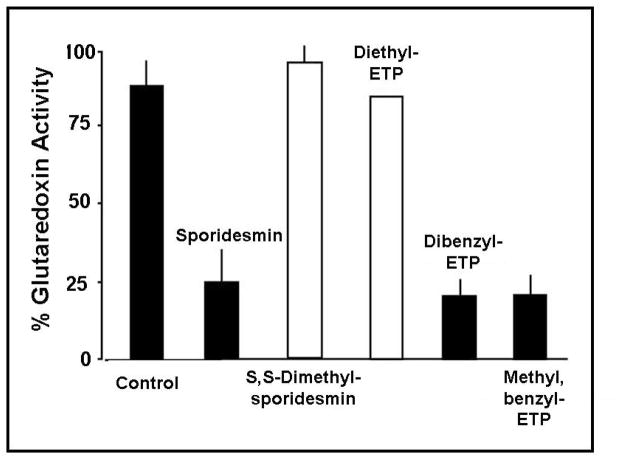
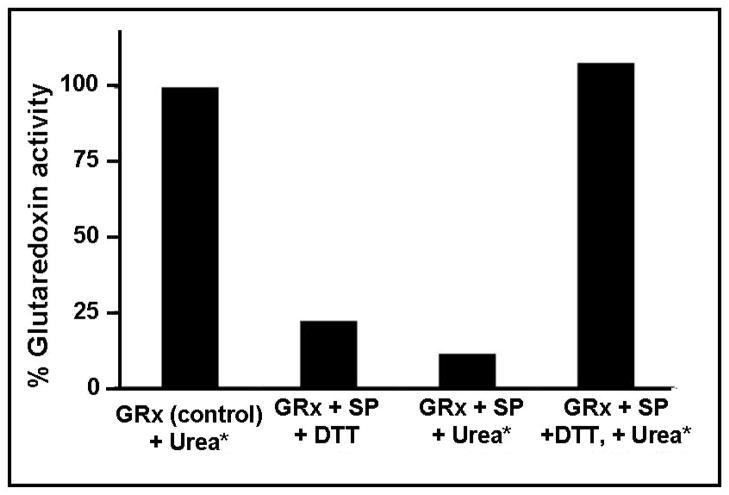
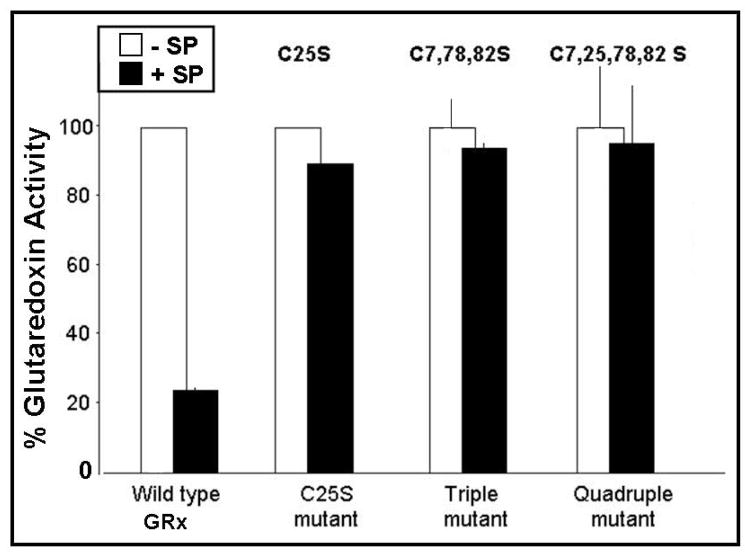
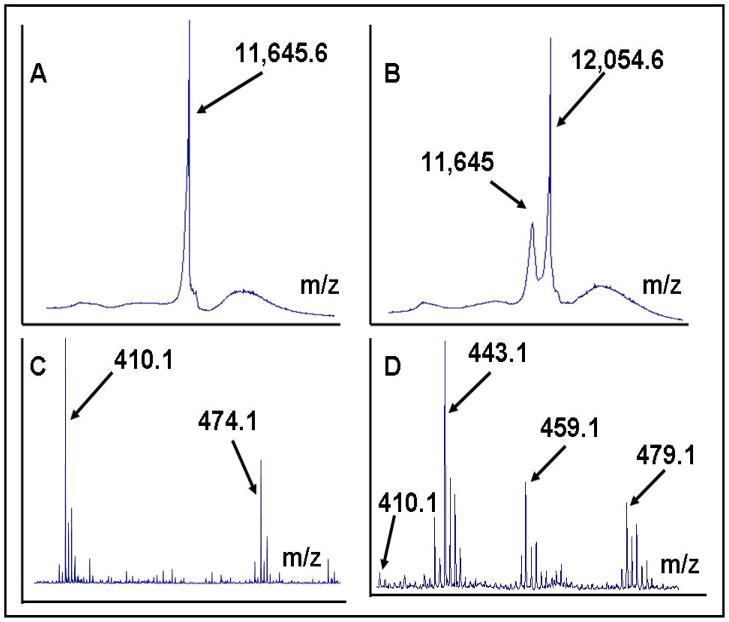
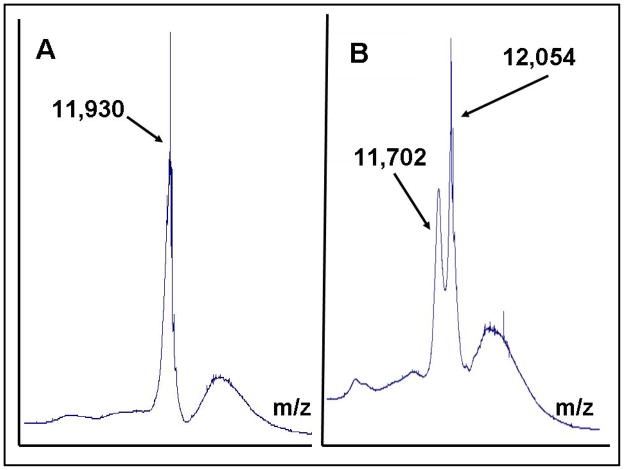
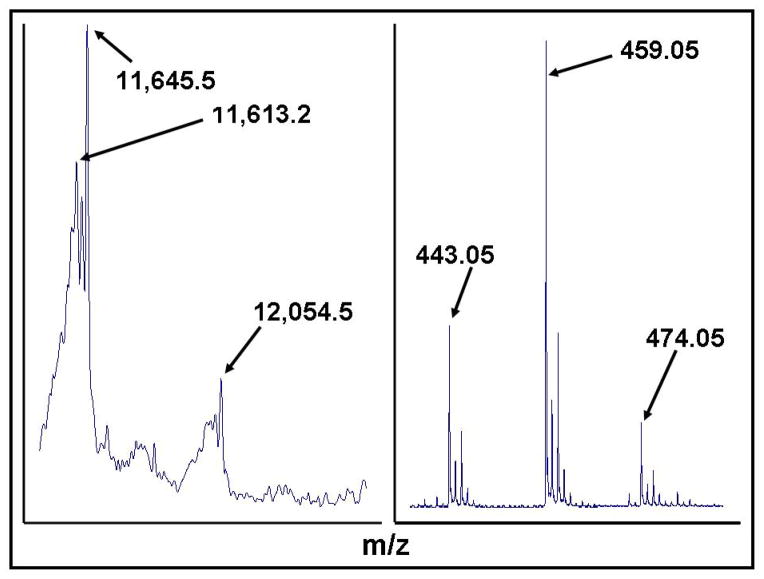
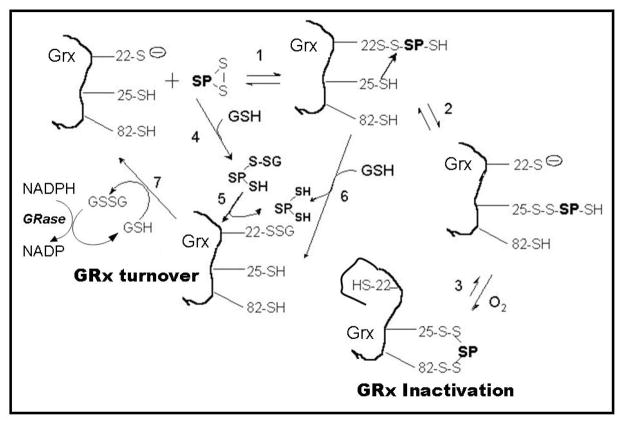
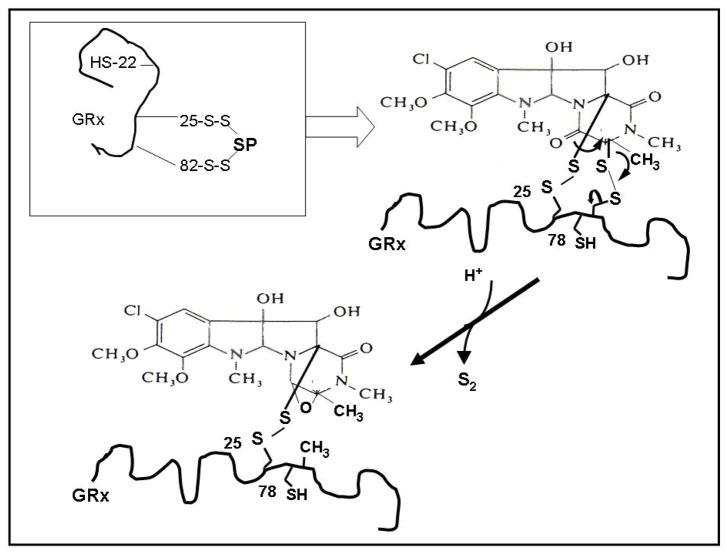
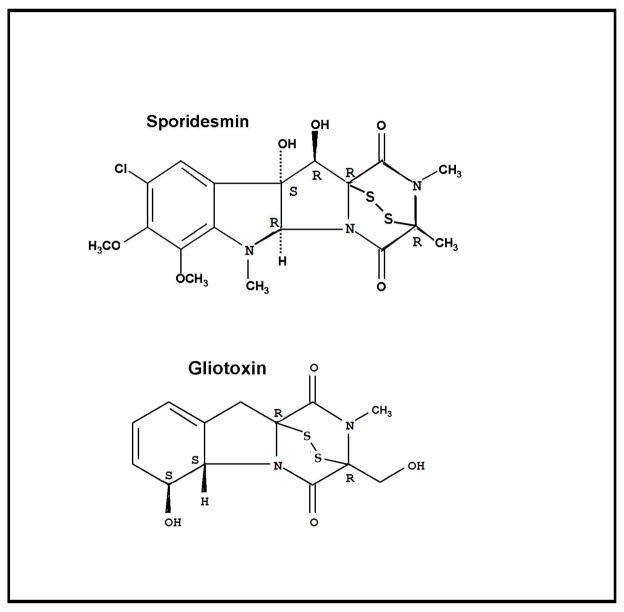
Glutaredoxin (thioltransferase) is a thiol-disulfide oxidoreductase that displays efficient and specific catalysis of protein-SSG deglutathionylation and is thereby implicated in homeostatic regulation of the thiol-disulfide status of cellular proteins. Sporidesmin is an epidithiopiperazine-2,5-dione (ETP) fungal toxin that disrupts cellular functions likely via oxidative alteration of cysteine residues on key proteins. In the current study sporidesmin inactivated human glutaredoxin in a time- and concentration-dependent manner. Under comparable conditions other thiol-disulfide oxidoreductase enzymes, glutathione reductase, thioredoxin, and thioredoxin reductase, were unaffected by sporidesmin. Inactivation of glutaredoxin required the reduced (dithiol) form of the enzyme, the oxidized (intramolecular disulfide) form of sporidesmin, and molecular oxygen. The inactivated glutaredoxin could be reactivated by dithiothreitol only in the presence of urea, followed by removal of the denaturant, indicating that inactivation of the enzyme involves a conformationally inaccessible disulfide bond(s). Various cysteine-to-serine mutants of glutaredoxin were resistant to inactivation by sporidesmin, suggesting that the inactivation reaction specifically involves at least two of the five cysteine residues in human glutaredoxin. The relative ability of various epidithiopiperazine-2,5-diones to inactivate glutaredoxin indicated that at least one phenyl substituent was required in addition to the epidithiodioxopiperazine moiety for inhibitory activity. Mass spectrometry of the modified protein is consistent with formation of intermolecular disulfides, containing one adducted toxin per glutaredoxin but with elimination of two sulfur atoms from the detected product. We suggest that the initial reaction is between the toxin sulfurs and cysteine 22 in the glutaredoxin active site. This study implicates selective modification of sulfhydryls of target proteins in some of the cytotoxic effects of the ETP fungal toxins and their synthetic analogues.
Figures
Similar articles
-
Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity.Biochemistry. 1998 Dec 8;37(49):17145-56. doi: 10.1021/bi9806504. Biochemistry. 1998. PMID: 9860827
-
Computational and mutational analysis of human glutaredoxin (thioltransferase): probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis.Biochemistry. 2006 Apr 18;45(15):4785-96. doi: 10.1021/bi0516327. Biochemistry. 2006. PMID: 16605247
-
Insights into deglutathionylation reactions. Different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the gamma-linkage present in glutathione.J Biol Chem. 2006 Nov 3;281(44):33107-14. doi: 10.1074/jbc.M605602200. Epub 2006 Sep 5. J Biol Chem. 2006. PMID: 16956877
-
Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system.Antioxid Redox Signal. 2004 Feb;6(1):63-74. doi: 10.1089/152308604771978354. Antioxid Redox Signal. 2004. PMID: 14713336 Review.
-
Thioltransferases.Adv Enzymol Relat Areas Mol Biol. 1993;66:149-201. doi: 10.1002/9780470123126.ch4. Adv Enzymol Relat Areas Mol Biol. 1993. PMID: 8430514 Review.
Cited by
-
Glutaredoxin: Discovery, redox defense and much more.Redox Biol. 2021 Jul;43:101975. doi: 10.1016/j.redox.2021.101975. Epub 2021 Apr 20. Redox Biol. 2021. PMID: 33932870 Free PMC article. Review.
-
Evaluation of a dithiocarbamate derivative as an inhibitor of human glutaredoxin-1.J Enzyme Inhib Med Chem. 2013 Jun;28(3):456-62. doi: 10.3109/14756366.2011.649267. Epub 2012 Feb 3. J Enzyme Inhib Med Chem. 2013. PMID: 22299579 Free PMC article.
-
Epidithiodiketopiperazines Inhibit Protein Degradation by Targeting Proteasome Deubiquitinase Rpn11.Cell Chem Biol. 2018 Nov 15;25(11):1350-1358.e9. doi: 10.1016/j.chembiol.2018.07.012. Epub 2018 Aug 23. Cell Chem Biol. 2018. PMID: 30146242 Free PMC article.
-
Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: implications for therapy of Parkinson's disease.Biochemistry. 2010 Mar 30;49(12):2715-24. doi: 10.1021/bi9018658. Biochemistry. 2010. PMID: 20141169 Free PMC article.
-
Self-protection against gliotoxin--a component of the gliotoxin biosynthetic cluster, GliT, completely protects Aspergillus fumigatus against exogenous gliotoxin.PLoS Pathog. 2010 Jun 10;6(6):e1000952. doi: 10.1371/journal.ppat.1000952. PLoS Pathog. 2010. PMID: 20548963 Free PMC article.
References
-
- Gravina SA, Mieyal JJ. Thioltransferase is a Specific Glutathionyl Mixed Disulfide Oxidoreductase. Biochemistry. 1993;32:3368–3376. - PubMed
-
- Mieyal JJ, Gravina SA, Mieyal PA, Srinivasan U, Starke DW. Glutathionyl Specificity of Thioltransferases: Mechanistic and Physiological Implications. In: Packer LEC, editor. Biothiols in Health and Disease. Marcel Dekker, Inc; New York: 1995. pp. 305–372.
-
- Jung CH, Thomas JA. S-Glutathiolated Hepatocyte Proteins and Insulin Disulfides as Substrates for Reduction by Glutaredoxin, Thioredoxin, Protein Disulfide Isomerase, and Glutathione. Arch Biochem Biophys. 1996;335:61–72. - PubMed
-
- Srinivasan U, Mieyal PA, Mieyal JJ. pH Profiles indicative of Rate Limiting Nucleophilic Displacement in Thioltransferase (Glutaredoxin) Catalysis. Biochemistry. 1997;36:3199–3206. - PubMed
-
- Yang Y, Jao S, Nanduri S, Starke DW, Mieyal JJ, Qin J. Reactivity of the Human Thioltransferase (C7S, C25S, C78S, C82S) Mutant and NMR Solution Structure of its Glutathionyl Mixed Disulfide Intermediate Reflect Catalytic Specificity. Biochemistry. 1998;37:17145–17156. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
